
Axonal mRNA localization and local translation in neurodegenerative diseases
2021-03-03 15:04JinXinLuYangWangYiJieZhangMeiFenShenHaiYingLiZhengQuanYuGangChen
中国神经再生研究(英文版) 2021年10期
Jin-Xin Lu , Yang Wang , Yi-Jie Zhang , Mei-Fen Shen Hai-Ying Li ,Zheng-Quan Yu , Gang Chen
Abstract The regulation of mRNA localization and local translation play vital roles in the maintenance of cellular structure and function. Many human neurodegenerative diseases,such as fragile X syndrome, amyotrophic lateral sclerosis, Alzheimer’s disease, and spinal muscular atrophy, have been characterized by pathological changes in neuronal axons,including abnormal mRNA translation, the loss of protein expression, or abnormal axon transport. Moreover, the same protein and mRNA molecules have been associated with variable functions in different diseases due to differences in their interaction networks.In this review, we briefly examine fragile X syndrome, amyotrophic lateral sclerosis,Alzheimer’s disease, and spinal muscular atrophy, with a focus on disease pathogenesis with regard to local mRNA translation and axon transport, suggesting possible treatment directions.
Key Words: Alzheimer’s disease; amyotrophic lateral sclerosis; axonal transport; fragile X syndrome; local translation; mRNA localization; neuron; spinal muscular atrophy
Introduction
The neuron is one of the most complex cells, with highly differentiated cell structures. Because axons and dendrites are located far from the nucleus, specialized methods for information transmission processing are often required to cope with local signal stimulation, and mRNA transport to axons and local mRNA translation are particularly important mechanisms. mRNA regulation is a key method through which cells regulate gene expression, converting genes to phenotypes. mRNA regulation occurs through three important mechanisms, including the regulation of mRNA localization,local translation, and stability, which are mediated by specific mRNA-binding proteins (Buchan, 2014).
The localization of mRNA to specific subcellular regions is very common, allowing genes to produce specific expression patterns, which can be spatially restricted within the cytoplasm.The local translation mechanism of mRNA is regulated by local signal stimulation, which protects the rest of the cell from some toxic products in other cellular compartments(Martin and Ephrussi, 2009). Several mechanisms determine the subcellular localization of mRNA: (1) motor proteins to transport mRNA to designated cell compartments along the cytoskeleton; (2) mRNA molecules can move through diffusion and capture by pre-positioned anchor protein; and(3) location-dependent mRNA degradation can control local mRNA levels (Medioni et al., 2012; Zappulo et al., 2017). The translation of mRNA into protein involves three sequential steps, including origination, extension, and termination(Hershey et al., 2019). Regulation can occur at any stage,but most regulation is largely limited to the initial stage of translation (Yan et al., 2017). Local translation in mammals has two primary functions. (1) It provides a means through which a neuron can respond to environmental signals and is involved in axon extension and pathfinding during axon development,in addition to synaptic and maintenance functions in mature cells. (2) Transcription factors and nucleocytoplasmic transport factors are often expressed through local translation, which can regulate transcription in the nucleus allowing axons to rapidly respond to damage and communicate with the cell body (Khalil et al., 2018). mRNA transport mechanisms play a substantial role in the subcellular of mRNA. For many genes, the transport of mRNA to various cell compartments is achieved by RNA-binding proteins (RBPs). RBPs with transport capabilities bind mRNA through the mutual recognition of cis-acting elements between specific RBPs and the cisregulating elements found in RNA molecules (Medioni et al.,2012) and then bind cytoskeleton motor proteins or adapters,facilitating mRNA transport (Czaplinski, 2014; Glock et al.,2017). These cis-acting elements are typically found in the 3′ untranslated region (3′UTR) of mRNA (Glock et al., 2017),and in some cases, the 3′UTR can even drive the positioning of proteins via mRNA translation. In axons, microtubules and micro-silks play important roles in the transport of mRNA,and mRNA molecules rely on cytoplasmic dynein and kinesin motors to reach their destinations (Guedes-Dias and Holzbaur,2019). The characteristics and numbers of active molecular motors recruited to the target mRNA determine the features of mRNA transport, including the type of cytoskeletal fibers used (actin filaments or microtubules), the type of movement(unidirectional or bidirectional), and the characteristics of mRNA movement (e.g., speed, progressive) (Medioni et al.,2012). Finally, during transportation, most transported mRNA appears to be in a dormant stage. In neurons, mRNA that has not reached its final destination must remain in a translationdormant state to prevent ectopic translation (Khan et al.,2015). The primary mechanism used to prevent premature translation is the use of a direct translation repressor, which binds to RNA regulatory sequences during RNA transport to prevent translation. This translation inhibition process primarily occurs at the initial stage (Besse and Ephrussi, 2008; Medioni et al., 2012).
Neurodegenerative diseases are clinically heterogeneous,including Alzheimer’s disease (AD), Parkinson’s disease(PD), fragile X syndrome (FXS), frontotemporal dementia(FTD), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD). These disorders include a range of similar symptoms and signs, including cognitive impairment, gait instability, weakness, emotional imbalance, and psychosis.Neurodegenerative diseases are progressive and eventually result in death due to primary central nervous system dysfunction or related medical complications. The incubation period from symptom onset to end-stage disease can range from several months to decades. One of the most striking features that is common to neurodegenerative diseases is the accumulation of endogenous proteins into misfolded,insoluble inclusions (Davis et al., 2018). Increasing evidence has indicated that local axon translation occurs in developing axons and in axons that respond to extracellular injury(Khalil et al., 2018). In many neurodegenerative diseases,abnormal changes occur in the transport, localization, and local translation of axonal mRNAs (Figure 1). The presence of ribosomal proteins, translation promoters, and ribosomal RNA has been reported in the axons of adult sensory neurons after injury (Koley et al., 2019). The rapid response to damage and the effective translation of mRNA into effector proteins typically requires the participation of RBPs. The ribonucleoproteins (RNPs) that are formed by the binding between RBPs and mRNA regulate all post-transcriptional stages, including localization and translation (Wang et al.,2016). The proper assembly and utilization of RNPs are essential for normal cell functions, and some studies have suggested that the hyper- or hypo-assembly of RNPs may cause neurodegenerative diseases, including spinal muscular atrophy (SMA) and ALS (Shukla and Parker, 2016). In addition,studies have suggested that the axonal transcriptome and proteome are altered in ALS and SMA (Baleriola and Hengst,2015). Some proteins and mRNAs have been specifically associated with certain disease presentations, such as Tau in AD, α-synuclein (ASN) in PD, and Cu-Zn superoxide dismutase 1 in ALS. These proteins have been found to be abundant in specific neurodegenerative diseases, whereas the expression of these proteins can barely be detected in other disease states. In addition, some proteins have been associated with multiple diseases. For example, sirtuins (SIRT1-SIRT7),TAR DNA-binding protein 43 (TDP-43), and heat shock proteins (HSPs) have been identified as being involved in the pathogenesis of AD, PD, and HD.
In this review, we provide an overview of four neurodegenerative diseases, ALS, SMA, AD, and FXS, with a focus on describing what is known about disease pathogenesis with regard to local translation and axon transport, and expound on the network of interactions between pathogenic factors.
Search Strategy
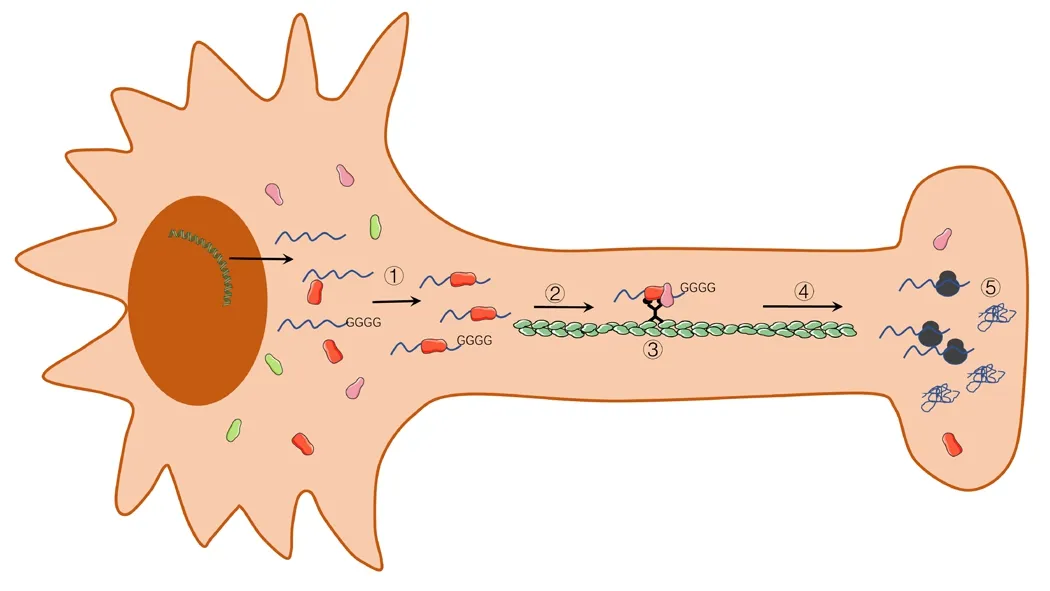
Figure 1|Abnormal mRNA transport, localization, and local translation in various diseases.
For the present review, we searched the literature using the following terms and words: “mRNA location” AND“neurodegenerative disease”; “mRNA location” AND“Amyotrophic lateral sclerosis” OR “ALS”; “mRNA location”AND “Alzheimer’s disease” OR “AD”; “mRNA location”AND “fragile X syndrome” OR “FXS”; “mRNA location” AND“spinal muscular atrophy” OR “SMA”; “local translation”AND “neurodegenerative disease”; “local translation” AND“Amyotrophic lateral sclerosis” OR “ALS”; “local translation”AND “Alzheimer’s disease” OR “AD”; “local translation”AND “fragile X syndrome” OR “FXS”; “local translation” AND“spinal muscular atrophy” OR “SMA”; “axonal transport”AND “neurodegenerative disease”; “axonal transport” AND“Amyotrophic lateral sclerosis” OR “ALS”; “axonal transport”AND “Alzheimer’s disease” OR “AD”; “axonal transport” AND“fragile X syndrome” OR “FXS”; “axonal transport” AND “spinal muscular atrophy” OR “SMA.” We performed a PubMed literature search of articles published during the period from January 2000 to June 2020 on mRNA location, axon transport in various neurodegenerative diseases, and interactions between molecules. In addition, we also used modifications of the above-listed keywords to thoroughly search the literature,such as “transport barriers” AND “neurodegenerative disease” OR “nervous system disease”; “mRNA transport”;“interaction” AND “neurodegenerative disease”; and nervous system disease. The results were further screened by title and abstract, and only studies examining rats, mice, humans,zebrafish, and nerve cells were included. The major inclusion criteria were literature pertaining to mRNA location, axon transport, and neurodegenerative disease.
A myotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is a neurodegenerative disease characterized by progressive muscle weakness, leading to palsy and death. One of the earliest pathological features of ALS is axonal retraction and degeneration (Akiyama et al., 2019). The most commonly associated mutations associated with ALS pathology have been identified in Cu-Zn superoxide dismutase 1, TDP-43, fused in sarcoma (FUS), and a hexanucleotide, amplified repeat of chromosome 9 in open reading frame 72 (López-Erauskin et al., 2018). Among these factors, TDP-43 and FUS have been the most widely studied proteins.
TDP-43 is an RBP, and pathogenic mutations have been shown to result in pathological aggregation, which is thought to drive the development of neurodegenerative diseases (Coyne et al.,2014). The insufficient axonal transport of TDP-43-targeted mRNA is thought to be associated with the pathogenesis of other neurodegenerative diseases, including ALS and FTD(Alami et al., 2014). TDP-43 binds to a specific RNA structure,known as the G-quadruplex, and TDP-43 has been shown to colocalize with G4 mRNA in neurites; therefore, TDP-43 is thought to facilitate the transfer of G4-containing mRNA to neurites, where the mRNA can be translated by local protein synthesis mechanisms (Ishiguro et al., 2016;Table 1). ALSrelated TDP-43 mutations impair the transport of mRNA to axons (Alami et al., 2014), which may be associated with the disruption of the microtubule cytoskeleton. Futsch, anin vivotarget molecule of TDP-43 (Coyne et al., 2014), has been shown to be indispensable for normal synaptic growth, which involves the synaptic microtubule cytoskeleton (Hummel et al., 2000; Roos et al., 2000). In Coyne’s experiment, TDP-43 was able to alter the axonal localization of futsch mRNA and protein, and TDP-43 has been shown to be involved in futsch mRNA transfer and translation at the neuromuscular junction (NMJ). In addition, futsch mRNA is inhibited by TDP-43 translation in motor neurons (Coyne et al., 2014). When TDP-43 is mutated, the synthesis of futsch becomes inhibited,and the normal function of the microtubule cytoskeleton is disrupted (Coyne et al., 2014). The microtubule cytoskeleton is important for the transport of mRNA to axons, and cytoskeletal disruptions may represent a pathological mechanism underlying the disrupted transport of mRNA to axons, contributing to ALS development. In another study,TDP-43 was found to regulate the transfer and translation of axonal mRNA and played a crucial role in the regulation of translation in dendrites during neuronal activity (Endo et al.,2018).
Mutations in the RBP FUS have also been associated with ALS. The localization of FUS is not unique, and FUS has been detected in both the nucleus and the cytoplasm (López-Erauskin et al., 2018), in addition to the presynaptic site and NMJ of hippocampal neurons (Thelen and Kye, 2019).Therefore, FUS appears to be active in neurons and may play a significant role in intracellular protein or mRNA transport.FUS has been reported to be involved in a number of cellular reactions, including transcription, splicing, RNA localization,and DNA damage response signaling (López-Erauskin et al.,2018; Naumann et al., 2018). The ALS-related mutation in FUS results in FUS accumulation along the axon, including near the local axonal translation site, triggering a local integrated stress response, which inhibits local protein translation and damages synaptic function (López-Erauskin et al., 2018;Table 1).Increasing levels of mutant FUS eventually result in reduced intra-axonal translation, leading to synaptic dysfunction(López-Erauskin et al., 2018).
TDP-43 and FUS do not function independently but have been shown to interact with each other, and ALS-related mutations in TDP-43 appear to promote this interaction (Birsa et al.,2020). Therefore, the co-occurrence of mutations in both TDP-43 and FUS is thought to result in the possible convergence of pathogenic pathways in ALS (Ling et al., 2010; Thelen and Kye, 2019). The pathological processes that underlie different diseases may overlap. Interactions have been identified between proteins associated with the pathogenic mechanisms of ALS and SMA, and direct interaction between FUS and survival of motor neuron (SMN) has been demonstrated,which can be influenced by FUS mutations. FUS and SMN colocalize in primary neurons, and the ALS-pathogenic FUS mutation results in the isolation of SMN into cytoplasmic aggregates, altering its subcellular distribution in cultured neuronal cells (Mirra et al., 2017). These results indicate that mutant FUS can impair local translation of axons by isolating the axon SMN-mRNA complex (Thelen and Kye, 2019).
Spinal Muscular Atrophy
SMA is an inherited disease that affects the motor neurons in the spinal cord and brain stem anterior horn, causing muscle atrophy and weakness (Bharucha-Goebel and Kaufmann,2017). Weakness in the proximal muscle tissue of SMA patients is an early feature associated with neurotransmission defects at the NMJ. The primary pathological feature of SMA is the loss of axons in the ventral roots and pathological changes at the NMJ (Courtney et al., 2019), followed by motor neuron degeneration. NMJ defects are the first pathological event that can be detected in an SMA mouse model (Ling et al., 2012; Goulet et al., 2013; Fallini et al., 2016). SMA has been associated with a mutation in SMN1, resulting in a decrease in the expression of SMN protein (Bowerman et al.,2017). The most characteristic function of the SMN complex is the assembly of small nuclear ribonucleoproteins (snRNPs),which are essential components of the pre-mRNA splicingmachinery (Pellizzoni, 2007). The complete deletion of Smn1 in mice results in early lethality (Schrank et al., 1997), and some studies have shown that SMA severity is correlated with the degree of impaired SMN complex activity in the spinal cords of SMA model mice (Gabanella et al., 2007). Therefore,understanding the effects of SMN mutations on SMN complex activity and function remain important goals.

Table 1 |Localization and local translation errors of different proteins in various diseases
A widely expressed protein, SMN plays a basic function in the assembly of a key component of the splicing bodysnRNPs (Fallini et al., 2016). Many studies have proposed that SMN plays a significant role in the modulation of mRNA localization and RBP regulation in neuronal axons (Fallini et al.,2016;Table 1). SMN appears to be involved in the regulation of axonal mRNA transport, stability, and local translation.Changes in the axonal localization of some mRNAs have been reported for some SMN-deficient cells, suggesting that SMN deficiency may result in the mislocalization of messenger ribonucleoproteins (mRNPs) required for axonal growth and function (Rihan et al., 2017). The localization of SMN,hnRNP, and β-actin to neuronal axons is very important for axonal outgrowth and function. Cultured primary motor neurons from SMA mice show the mislocalization of β-actin mRNA(Gabanella et al., 2007), resulting in axonal growth disorder through a mechanism that is regulated by human antigen D (HuD) (Rathod et al., 2012). HuD is a member of the embryonic lethal abnormal vision protein family that binds to the AU-rich elements in the 3′UTR of target mRNA molecules to modulate mRNA stability and translation (Fallini et al., 2011). HuD regulates multiple stages after mRNA transcription, including stability, alternative splicing, RNA localization, and translation (Tebaldi et al., 2018). The effects of HuD on SMN and the extent of their interactions have become a focus of SMA research. Studies have suggested the existence of a biochemical link between SMN and HuD,and researchers have shown that these two proteins are components of a complex that is actively transferred along motor neuron axons (Fallini et al., 2011). Moreover, SMN defects may affect HuD transport to motor neurons axon by affecting mRNP assembly and the transport of mRNP complexes that contain HuD (Fallini et al., 2011), affecting the axonal localization of HuD. Moreover, SMN has been shown to interact directly with HuD, promoting the binding between HuD, mRNA, and other proteins in the mRNP complex (Fallini et al., 2011). Weakened SMN-HuD interactions have also be observed in SMA patients (Fallini et al., 2011).
In addition, growth-associated protein 43 (GAP43) is highly expressed during neuronal growth and axonal regeneration and is involved in the regulation of actin cytoskeleton dynamics (Hartl and Schneider, 2019). Another study identified GAP43 as a novel target for SMN (Fallini et al.,2016). SMN can regulate the axonal localization of GAP43 and the transcription of candidate plasticity-related gene 15(cpg15) (Wang et al., 2016). GAP43 mRNA and protein levels were found to be reduced in the axons and growth cones of motor neurons in an SMA model mouse (Fallini et al.,2016). CPG15 protein plays several important roles in neural development. The expression of CPG15 is spatiotemporally correlated with synaptic growth, activity-dependent plasticity,and neuronal migration (Zhao et al., 2017). The deletion of SMN results in decreased cpg15 mRNA expression levels, and cpg15 overexpression was able to partially rescue an SMNdeficiency phenotype in SMA model zebrafish (Akten et al.,2011). Previously, cpg15 was reported to be involved in the promotion of neuronal growth and dendritic arbor growth,which may represent another mechanism through which SMN loss may result in axonal abnormalities (Zhao et al., 2017).The overexpression of HuD and insulin-like growth factor 2 messenger RNA binding protein (IMP1) were found to be sufficient to restore GAP43 levels at the growth cone and were able to rescue axonal growth defects in SMA motor neurons(Fallini et al., 2016). Imp1 is an RNA-binding protein that plays a role in the localization and local translation of some mRNAs and can rescue the motor neuron axon impairment observed in SMA models (Fallini et al., 2016). Finally, SMN has been associated with the regulation of protein arginine methyltransferase 4 (PRMT4) translation, which is also known as coactivation-related arginine methyltransferase 1(CARM1) (Sanchez et al., 2013; Singh et al., 2017). CARM1 is a multifunctional protein that affects transcription and splicing.The reduction of SMN can increase the level of CARM1(Sanchez et al., 2016). SMN protein has also been shown to interact with many other disease marker proteins, including fragile X mental retardation protein (FMRP) in neuronal mRNP granules (Piazzon et al., 2008) and the ALS-related proteins FUS/TDP43 through a common pathway (Yamazaki et al.,2012; Guo et al., 2017).
These findings suggest that in SMA patients, the lack of SMN results in the abnormal expression of various mRNAs, resulting in abnormal axonal phenotypes.
Alzheimer’s Disease
Alzheimer’s disease (AD) is the most widespread neurodegenerative disease, characterized by a gradual loss of cognitive and functional abilities (Khalil et al., 2018). The microscopic examination of the AD brain has revealed the coexistence of two types of abnormal structures: extracellular amyloid plaques, formed by β-amyloid peptide (Aβ); and neurofibrillary tangles (NFT), composed of tau. Both of these structures contain highly insoluble, densely packed fibers(Bloom, 2014).
In the healthy brain, tau is a unique axonal protein that is involved in the formation and stability of microtubules. In the AD brain, tau protein is hyperphosphorylated and forms into fibers, which appear as neurofilaments in dendrites and as NFTs in dendritic cells and axons. Tau exhibits a pre-tangle state before NFTs are formed; in this state,hyperphosphorylated and non-fibrotic tau accumulates in the neuron cell body and axon (Kobayashi et al., 2017). In another experiment, tau hyperphosphorylation and misfolding were found to be significantly increased in AD (Tai et al., 2014),resulting in the formation of NFTs and accelerating brain damage associated with AD.
The structure of tau is well-known to be very important for its function. Although it is beyond the scope of this review to introduce the structure of tau in detail, the function of the tau protein should be discussed. The N-terminal domain of tau is distal from microtubules and does not interact directly with microtubules; however, the N-terminal domain of tau is known to participate in the regulation of microtubule dynamics and affects the connection between microtubules and other cellular components (Guo et al., 2017;Table 1).Some studies have shown that hyperphosphorylated tau displays reduced interactions with microtubules, which may lead to the loss of axon-specific tau localization, triggering the pathological AD cascade (Gauthier-Kemper et al., 2011).Tau is typically selective localized to the axonal compartment(Gauthier-Kemper et al., 2018) and is present at very low concentrations in dendrites, including dendritic spines (Guo et al., 2017). Tauopathies are associated with the mislocalization of tau. Previous studies have shown that glutamate can stimulate the rapid local translation of tau mRNA in dendrites.Glutamate is an important excitatory neurotransmitter that can activate ionic [including N-methyl-D-aspartic acid (NMDA)and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid]and metabolic glutamate receptors, and glutamate receptor activation has been associated with the rapid translation of tau mRNA in dendrites (Kobayashi et al., 2017). When the glutamate content changes, the tau contents also change rapidly, resulting in a cascade of subsequent reactions. Studies have shown that tau protein is involved in the regulation of the neurite maturation signaling pathway and may be involved in morphological plasticity in response to brain-derived neurotrophic factors (Gauthier-Kemper et al., 2011).
β-amyloid (Aβ) is an important contributor to AD pathology,and Aβ1-42is considered to be an important cause of most of the neurodegenerative changes associated with AD(Baleriola et al., 2014). Aβ is obtained from amyloid precursor protein through sequential cleavage events catalyzed by βand γ-secretase (Wang et al., 2017). The increased rate of pathological amyloid precursor protein cleavage in AD results in increased Aβ concentrations and the phosphorylation of tau protein (Gauthier-Kemper et al., 2011). In addition, the oligomeric form of Aβ activates signaling pathways containing Fyn, a mitogen-activated protein kinase/extracellular signalregulated kinase (MAPK/ERK), and S6, which causes the local translation of tau mRNA in dendrites. The accumulation of Aβ and tau in the AD brain appear to be linked by the kinase Fyn(Li and Götz, 2017). The presence of Aβ1-42in the axon triggers the recruitment of specific mRNAs, including activated transcription factor 4 (ATF4), a protein that is locally translated in the axon, is activated in many neurodegenerative diseases,including AD, and is known to be involved in the unfolded protein response (UPR) pathway (Baleriola et al., 2014).
Although the link between AD and disrupted local axonal translation is not as clear as has been described for other diseases, the insoluble aggregates found in the brains of AD patients are rich in U1-70k and other U1 snRNP spliceosome components, indicating that the dysregulation of RNP assembly and RNA splicing may contribute to AD pathogenesis (Khalil et al., 2018). In addition, some studies have demonstrated that TDP-43 may be related to the expression of tau (Amador-Ortiz et al., 2007). Although the role played by TDP-43 in AD progression is not obvious, the pathological overlap between TDP-43 and tau will help us further understand the underlying disease mechanism (Khalil et al., 2018).
Fragile X Syndrome
FXS is caused by the loss of FMRP, which is an RBP that binds many mRNAs. FMRP often inhibits mRNA translation and participates in other mRNA control modes, such as trafficking FMRP-associated mRNA granules from the soma to neurites(Ifrim et al., 2015; Hsu et al., 2019). Many of the mRNAs regulated by FMRP encode proteins that control synaptic structure and function, such as postsynaptic density (PSD)-95, Arc, and Shank1 (Banerjee et al., 2018). FMRP inhibits the translation of mRNA, preventing basic or activity-dependent local protein synthesis (Bagni and Oostra, 2013) by affecting mRNA stability, in combination with RNA-induced silencing complex (RISC; DeMarco et al., 2019). Typically, the loss of FMRP protein is caused by the repeated amplification of a trinucleotide repeat (CGG) in 5′UTR of FMR1. When the number of trinucleotide repeats exceeds over 200 copies,this region becomes methylated, together with the FMR1 promoter, leading to transcriptional inhibition (Thelen and Kye, 2019).
At the cell morphology level, the loss of FMRP function in FXS results in the pathological hyperabundance of long,thin, immature dendritic protrusions (Khayachi et al., 2018);therefore, the abnormal regulation of dendritic spine plasticity may be an underlying pathological mechanism associated with this disease. Actin is known to be critically involved in the regulation of neuronal polarization and synaptic plasticity(He et al., 2016). Therefore, the phenotypic characteristics of FXS may be caused by a kinetic imbalance of activitydependent actin assembly in the dendritic spine (Feuge et al., 2019). However, FMRP is the key protein involved in the development of FXS. Although hundreds of mRNAs have been identified to interact with FMRP, actin-binding protein (ABP) mRNAs that directly interact with FMRP are rare (Feuge et al., 2019). In the absence of FMRP, thousands of neuronal proteins become dysregulated (Tabet et al.,2016). Experiments using cross-linking immunoprecipitation to detect homologous targets that interact with FMRP have suggested that diacylglycerol kinase kappa (DGK-κ), which is expressed widely throughout the brain, is the most efficient mRNA for the cross-linking immunoprecipitation of FMRP.The loss of DGK-κ leads to the FXS phenotype; however,whether DGK-κ participates in synaptic plasticity remains unclear (Tabet et al., 2016). In addition, FMRP can regulate the transport of specific mRNAs to synapses. Previous studies have demonstrated that FMRP interacts with Ca2+/calmodulindependent protein kinase II mRNA to promote the localization of the mRNA in dendritic spines and promote translation(Bagni and Oostra, 2013). Following stimulation with (S)-3,5-dihydroxyphenylglycine (DHPG), which can induce a synaptic plasticity state called long-term depression (DHPG-induced long-term depression), FMRP interacts with motor proteins on microtubules to promote the activity-dependent localization of mRNA to the synaptic spine (Bagni and Oostra, 2013). In the absence of FMRP, FMRP-targeted mRNAs (such as Map1b and Sapap4) appearing to mislocalize, which likely directly affects the abnormality of the synaptic structure, disrupting signal transmission (Dictenberg et al., 2008).
In addition, FMRP affects the stability of mRNA. PSD-95 mRNA a component of the FMRP-mRNP complex in vivo, and FMRP has been shown to interact directly with the 3′UTR of PSD-95 mRNA. The interaction between FMRP and PSD-95 mRNA appears to promote the stability of PSD-95 mRNA, which can be further enhanced by metabotropic glutamate receptor(mGluR) stimulation. However, in FMRP1 knockout mice, PSD-95 mRNA was less stable (Zalfa et al., 2007). Previous studies have demonstrated that although synapses can form in the absence of PSD-95, the process of establishing a functional network becomes abnormal, which may lead to abnormal changes in dendritic spine density (Vickers et al., 2006; Coley and Gao, 2019). Therefore, FMRP may influence NMDA and mGluR receptor signaling by regulating various proteins,including PSD-95, and the destruction of this complex may cause the cognitive defects observed in FXS (Zalfa et al., 2007).
Interactions within a Network
We have identified a number of proteins that play important regulatory roles in several diseases.
Sirtuins (SIRT1-SIRT7) belong to the histone deacetylase family. These enzymes regulate the properties and functions of various proteins, including histones, kinases, and transcription factors (TFs), by removing acetyl groups that have been post-translationally attached to lysine residues by acetyltransferases (Jęśko and Strosznajder, 2016; Kupis et al., 2016; Jęśko et al., 2017). Due to this unique mode of action, sirtuins have been identified to interact with proteins associated with AD, PD, HD, and ALS. SIRT1 has been reported to alter the balance between APP amyloidosis and nonamyloidosis, both in vitro and in transgenic mouse models (Qin et al., 2006), Sirtuins can also mediate the leptin-dependent inhibition of tau phosphorylation (Greco et al., 2011). In addition, SIRT1 and tau share a common upstream regulatory mechanism, as they are both miRNA-132 and AMP-activated kinase (AMPK) targets (Hernandez-Rapp et al., 2016; Jęśko et al., 2017). In PD patients, SIRT1 may play a protective role,through several stress-reducing pathways, especially to ASN metabolism (Jęśko et al., 2017). Hsp70 has also been shown to prevent ASN aggregation and toxicity (Klucken et al., 2004;McLean et al., 2004). SIRT1 also deacetylates heat shock factor 1 (HSF1), facilitating the prolonged binding between SIRT1 and the Hsp70 gene-coding target sequence (Westerheide et al.,2009), which further affects the function of ASN in PD. HSF1 and Hsp70 may also mediate the protective effects of SIRT1 in the ALS model by reducing the misfolding and aggregation of Cu/Zn-superoxide dismutase (Watanabe et al., 2014). In HD disease, sirtuins play roles in neuronal survival and are known to interact with Huntingtin protein (Jiang et al., 2011)and forkhead box class O (FOXO) TFs (Jęśko and Strosznajder,2016), indicating that they may be possible research targets(Jęśko et al., 2017).
Some proteins may play similar roles in multiple diseases.In ALS, TDP-43 has been shown to play a pathogenic role by participating in the nuclear output of RNA, the axonal transport of mRNA, and regulating the cytoskeleton. In AD,TDP-43 also plays an important role and may be involved in the regulation of tau expression (Amador-Ortiz et al.,2007). HSPs are among the most phylogenetically conserved superfamilies of chaperones and are known to play active roles in a variety of neurodegenerative diseases. Studies have shown that the overexpression of HSPs can prevent the aggregation and associated toxicity of several disease-related proteins (Yerbury et al., 2016), including Aβ and tau protein in AD (Lackie et al., 2017), polyglutamine-expanded proteins in HD (Ormsby et al., 2013), α-synuclein in PD (Cox et al., 2014),and superoxide dismutase 1 in ALS (Novoselov et al., 2013;Figure 2).
Few studies have examined the interactions among pathogenic factors that are shared across different neurodegenerative diseases; therefore, the development of a complete network remains challenging, and further research in this field is necessary to develop these networks further.
Conclusions
In this review, we reviewed several methods of mRNA regulation, including the regulation of mRNA localization,translation, and stability. We also explored the importance of these mechanisms and the contributions of local translation and axon transport abnormalities to the development of various neurodegenerative diseases, including ALS, SMA,FXS, and AD (Table 1). We mainly explored the pathogenic mechanisms associated with these diseases and the interactions between different pathogenic factors.
Current research has revealed the importance of mRNA regulation during neural development. We believe that mRNA regulatory mechanisms offer unique advantages that can be used to fine-tune gene expression. First, mRNA regulatory mechanisms can directly guide a specific protein to the correct intracellular compartment while preventing it from being expressed elsewhere and protecting other components of the cell from damage. Second, these mechanisms allow synapses to limit gene expression at the temporal and spatial levels. Therefore, activated synapses can initiate local protein translation, which can alter the function and morphology of the surrounding microenvironment (Doyle and Kiebler, 2011; Avnet et al., 2019). This control method is very important, allowing neurons to respond rapidly to environmental stimuli. During neurodegenerative diseases,mRNA localization and local translation have been shown to have significant effects. This review focused on the effects of several disease-associated proteins on diseases, including HuD, FMRP, and TDP-43. Most of these proteins are known to participate in the local translation of mRNA, and some are specifically active during the course of specific diseases. For example, FXS is primarily caused by the deficiency of FMRP(Ifrim et al., 2015). In contrast, some proteins are involved in the pathogenic processes of many diseases, such as TDP-43.Studies have shown that insufficient mRNA axonal transport targeted by TDP-43 may contribute to the pathogenesis of related diseases, such as ALS and frontotemporal dementia(Alami et al., 2014), and in AD, TDP-43 has been confirmed to be involved in the expression of the AD marker protein tau (Amador-Ortiz et al., 2007). In addition, extracellular stimulation signals may also affect the underlying mechanisms associated with these diseases. Some studies have confirmed that changes in neurotrophic factor signaling may be involved in the progression of neurodegenerative diseases and other psychiatric diseases (Villarin et al., 2016). Although we have a deep understanding of the mechanisms through which axon transport and local translation occur, many of the related mechanisms and their contributions to the pathogenesis of neurodegenerative diseases remain unclear. In recent years,although we have used increasingly sensitive and advanced methods to identify axon transcriptomes and identify abnormally expressed proteins associated with disease states,the impacts of these transcriptomes and proteins on disease development remain poorly understood.
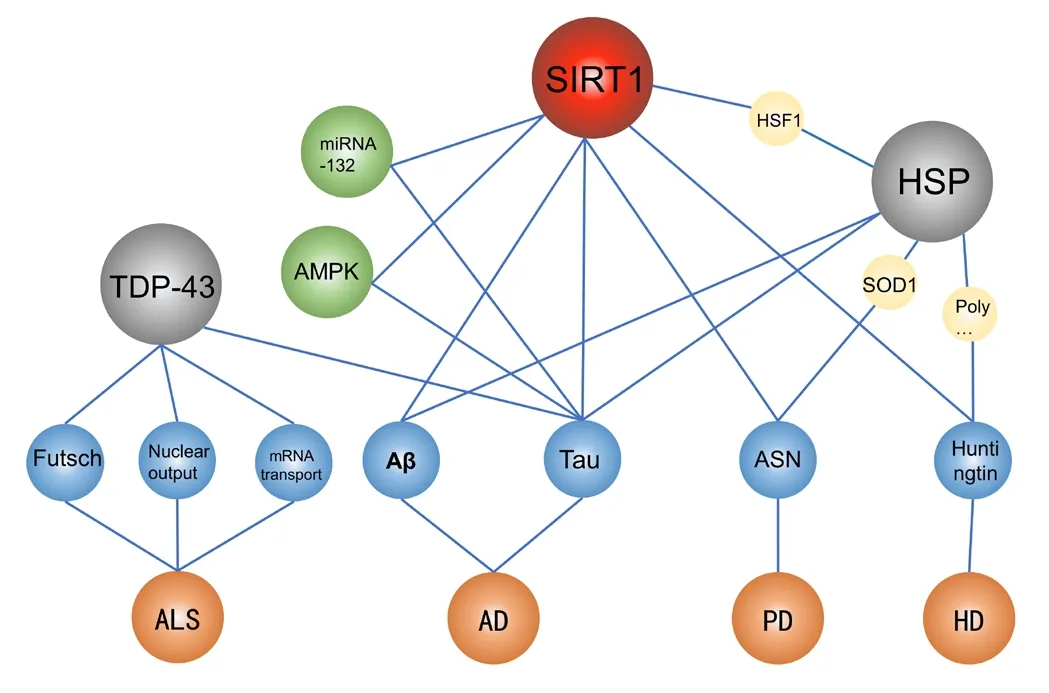
Figure 2|The interactions among disease-associated proteins.
Therefore, the pathogenic contributions of these changes in axonal transport and local axonal translation require additional research. For the diseases described above, the continued exploration of significant pathogenic mechanisms remains necessary. We must continue to identify common mechanisms associated with neurodegenerative diseases, to study these problems with a unified vision, and explore the various factors that contribute to disease states to develop feasible methods for disease treatment and prevention.
Acknowledgments:We acknowledge all the staff of Department of Neurosurgery & Brain and Nerve Research Laboratory, The First Affiliated Hospital of Soochow University for their support.
Author contributions:Review conception and design: GC, ZQY, HYL;manuscript writing: JXL, YW; literature retrieval: YJZ; manuscript review and editing: MFS. All authors read and approved the final manuscript for publications.
Conflicts of interest:All authors declare that there is no conflict of interests.
Financial support:This work was supported by the National Natural Science Foundation of China, Nos. 81830036 (to GC), 81771255 (to GC), 81771254(to HYL), 81971106 (to ZQY); Project of Jiangsu Provincial Medical Innovation Team, No. CXTDA2017003 (to GC); Jiangsu Provincial Medical Youth Talent,No. QNRC2016728 (to HYL); the Natural Science Foundation of JiangsuProvince, No. BK20170363 (to HYL); Gusu Health Personnel Training Project,No. GSWS2019030 (to HYL).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Axonal regeneration and sprouting as a potential therapeutic target for nervous system disorders
- The role of gap junctions in cell death and neuromodulation in the retina
- Don’t know what you got till it’s gone: microglial depletion and neurodegeneration
- Low-dose lipopolysaccharide as an immune regulator for homeostasis maintenance in the central nervous system through transformation to neuroprotective microglia
- Protein post-translational modifications after spinal cord injury
- Alzheimer’s disease: a tale of two diseases?