
富锂锰基正极材料研究进展
2021-03-02 13:17:18南文争王继贤彭思侃燕绍九
航空材料学报 2021年1期
南文争, 王继贤, 陈 翔, 彭思侃, 燕绍九*
(1.中国航发北京航空材料研究院,北京 100095;2.北京石墨烯技术研究院有限公司,北京 100095)
锂离子电池因具有高比能量、无记忆效应和长寿命等优点,被认为是最具潜力的电化学储能器件之一。随着社会的发展,电动汽车、消费类(3C)电子产品和储能装置等对锂离子电池的能量密度提出了更高要求[1-2]。以电动汽车为例,续航里程短是其推广应用中存在的主要难题。目前,欧美、日本和中国都制定了明确的电池路线图,即在未来五到十年内,将锂离子电池单体的能量密度从现在的200~250 Wh/kg提高到300~500 Wh/kg。因此,发展高比容量、高电压正极材料以提升电池能量密度成为研究热点。
目前商业化应用的正极材料主要有锰酸锂(LiMn2O4)、磷酸铁锂(LiFePO4)、钴酸锂(LiCoO2)、镍钴锰酸锂(LiNixCoyMn1-x-yO2)及镍钴铝酸锂(LiNixCoyAl1-x-yO2)[3]。上述材料的实际比容量为110~200 mAh/g[4-5](表1)。相比于商业化负极材料(石墨负极比容量为372 mAh/g,硅基负极比容量为2000 mAh/g)[6],正极材料能量密度提升已成为锂离子电池发展的关键因素[7]。富锂锰基正极材料(Li2MnO3•LiNixCoyMn1-x-yO2)凭借高比容量(≈ 250 mAh/g)、高工作电压(≈ 3.6 V)及低成本等优势[8],有望成为下一代商用高比能锂电池的正极材料[9-10],已受到学术及工程界的极大关注。
虽然作为锂离子电池正极材料具有巨大潜力,但富锂锰基正极材料也存在诸多问题,如倍率性能差、首次库仑效率低及电压/容量衰减快等[11],限制了其工程化应用。为此,国内外学者们开展了大量工作,并取得一系列重要进展。研究发现,材料主要失效机制包括晶格氧析出、过渡金属离子迁移、电极/电解液界面副反应等。离子掺杂、表面包覆和晶体结构调控等技术手段可显著改善富锂锰基正极材料的电化学性能。
本文从材料结构、电化学反应机理、失效机制及改性方法等几方面阐述了富锂锰基正极材料近年来的研究进展,基于目前的实验进展及理论认知,对材料未来的研究方向进行了展望。
1 富锂锰基正极材料结构及电化学反应机理
1.1 结构特点
表1为几种典型正极材料的电化学特征[12-17]。富锂锰基正极材料由LiMO2[18](M= Co、Mn、Ni等)和Li2MnO3两种组分构成,分子式写作xLi2MnO3•(1-x)LiMO2。两种组分结构相似,均为α-NaFeO2类型的层状结构,其中氧原子呈立方密排方式排列。LiMO2结构中的过渡金属(TM)层不含Li+,属于六方晶系空间群(图1(a));而Li2MnO3结构中过渡金属层中的Mn有三分之一被Li取代(图1(b)),形成Li被六个Mn所包围的“蜂窝”结构(图1(c))[19]。这种有序的Mn、Li排列形成了LiMn6超晶格结构,正因该结构的存在,使得Li2MnO3的点群对称性由变为单斜晶系C2/m。因此,Li2MnO3可看成LiMO2结构的特殊形式[20],其分子式可用类似于LiMO2结构的分子式表示为Li[Li0.33Mn0.67]O2。此外,LiMO2相(001)晶面间距与Li2MnO3相(003)晶面间距接近(≈0.47 nm),两相具有形成固溶体的可能性。正因两者结构的高度相似性,学术界对富锂锰基正极材料结构的认识仍存在争议[21-23]。

表 1 几种典型正极材料的电化学特征Table 1 Electrochemical characteristics of typical cathode materials

图 1 富锂锰基正极材料结构示意图[19] (a)LiMO2相;(b)Li2MnO3相;(c)Li2MnO3相中过渡金属层的原子排布Fig. 1 Structural diagrams of Li- and Mn-rich cathode materials[19] (a) Li2MO3 phase; (b) Li2MnO3 phase; (c) atoms ordering within Li(TM)2 layer of Li2MnO3 phase
一些学者认为富锂锰基正极材料为LiMO2相及Li2MnO3相的复合物。Mohanty等[24]通过中子衍射对Li1.2Mn0.55Ni0.15Co0.1O2材料进行表征,将得到的数据进行精修分析,图谱中既有结构特征峰,也有C2/m结构特征峰。若将材料看成由50%六方LiMO2相和50%单斜Li2MnO3相组成的复合结构,则得到的数据与实验结果一致,他们进一步采用磁性分析方法确认了富锂锰基正极材料为两相复合结构。Yu等[25]通过高分辨透射电镜(HRTEM)和电子能量损失谱(EELS)发现,Li1.2Mn0.567Ni0.166Co0.067O2材料由LiMO2和Li2MnO3两相组成,直接证明其为两相复合物。
1.1.3 单斜C2/m单相结构相结构。Jarvis等[18]采用XRD对Li[Li0.2Ni0.2
还有一些学者认为富锂锰基材料属于C/2m单Mn0.6]O2材料进行分析,从表征结果得出了两种结论:(1)材料是由相、C2/m相组成的复合结构;(2)材料属于C2/m单相结构。他们采用NBED及HAADF-STEM对结构进行进一步分析,结果没有显示两相复合结构的存在。由此得出结论,材料为C2/m单相结构。此外,Genevois等[28]利用HAADFSTEM及中子衍射证明Li1.2Co0.13Ni0.13Mn0.54O2材料为C2/m单相结构,该结构中长程有序的Li2MnO3被许多无序LiMO2区域分离。
虽然存在上述争论,但大多数研究者认同材料的晶体结构与组分、合成方式及热处理制度等密切相关。这也很好解释了目前关于富锂锰基正极材料具有不同结构认识的原因。有研究认为,Li1.2Co0.4Mn0.4O2材料在2~3 nm范围可看成Li2MnO3和LiCoO2的局域复合相[29]。在材料合成过程,如果冷却速度过慢,局域的两相晶粒有充足时间长大,并扩展到整个晶体结构中,形成明显晶界,最终导致两相从固溶体结构中分离出来形成复合结构,并出现尖晶石结构[30]。另有研究认为,富锂锰基正极材料中的Mn可维持材料结构稳定,但Mn比例过高将破坏LiMO2结构,使之向尖晶石结构转变[31];Ni可提高材料可逆嵌锂容量,但Ni比例过高会造成Li、Ni混排,阻碍Li+传输,进而影响材料性能[32]。
综上,不管富锂锰基正极材料整体结构是单一固溶体还是两相复合物,其局域结构是复杂的,而局域结构对材料整体结构的定性及材料的电化学性能都有着重大影响。为获得高性能富锂锰基正极材料,优化材料合成路线和保证元素的均一分布等尤为必要。此外,研究者应利用先进表征手段揭示材料合成及循环中的结构演变,深化对材料结构的认识。
1.2 电化学反应机理
富锂锰基正极材料具有高的放电比容量(>250 mAh/g)[33-34],不同于传统层状材料,其充放电过程不仅包含过渡金属离子的氧化还原反应,还涉及氧阴离子的电荷补偿反应[35-36]。
0.5Li2MnO3•0.5LiMn0.42Ni0.42Co0.16O2材料的首次充电曲线包含两段:一个短的电压斜坡段(< 4.5 V)和一个长的电压平台段(≈ 4.5 V)。“斜坡”表示Li+从LiMO2组分中脱出,伴随Ni2+、Co3+的氧化反应(Mn4+价态不变)。当材料中Ni2+和Co3+全部转变为+4价,电压接近4.5 V[37]。该过程不同于传统层状材料,Li+可全部从LiMO2结构中脱出,是因为Li2MnO3组分起到了稳定材料结构的作用。关于材料长电压平台段,Li等[38]率先提出“氧丢失”机理进行了解释:该阶段Li+从Li2MnO3结构中脱出,实质为Li和晶格氧同时脱出,形式上相当于脱出了“Li2O”[39]。首次充电过程可用以下反应式表示[40-41]:
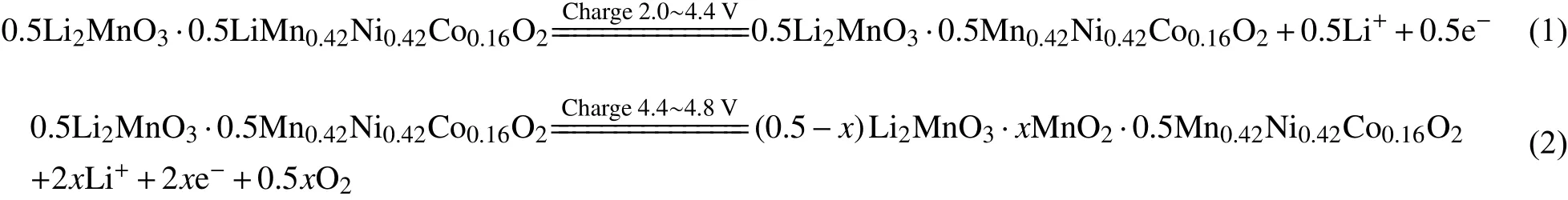
首次放电过程,当放电电压高于3.5 V时,发生可逆的电化学还原反应,即(1)过程脱出的Li+全部回嵌到正极材料中,伴随过渡金属离子价态的升高;当放电电压低于3.5 V,(2)过程脱出的Li+部分回嵌至材料晶格,发生不可逆还原反应。总反应式可表示如下[40-41]:

根据上述反应,得出材料理论放电比容量为261 mAh/g,然而不少研究报道的富锂锰基正极材料实际容量远高于该值,因此上述“氧丢失”机制仍不能清晰解释材料具有高容量的原因。Orishi等[42]和Han等[43]对该机理进一步完善,认为表面和体相的晶格氧发生了不同的氧化还原反应。通过系列研究发现,首次充电过程,材料中O原子的变化可分为两部分:一部分失电子生成氧气释放,伴随过渡金属离子迁移而发生结构重排;另一部分通过形成局部空位电子的方式发生可逆氧化还原反应,形成类过氧根基团(2O2-/O22-),该过程不涉及氧气释放及结构转变。
综上,Li2MnO3相的存在是富锂锰基正极材料具有高容量的根本原因,同时Li2MnO3相的活化也引发了一系列问题,制约了材料的产业化发展。目前富锂锰基正极材料的电化学反应机理尚不明确,材料的微观结构及电化学性能间的构效关系有待更深入、系统的研究,以为工程化应用提供理论支撑。
2 失效机制
2.1 晶格氧析出
在富锂锰基正极材料结构中,存在Li—O—Li局部配位结构,该构型中的O2p轨道电子会流向过渡金属的空d轨道来降低整个系统自由能,形成的亚稳态氧通过形成类过氧根形式的物质(O2n-)得到稳定[44]。电极发生电化学反应时转移的电子来自非键状态的O2p轨道,这种同过渡金属离子没有结合的轨道电子的转移并不直接破坏材料结构,因此通过该反应机制(还原耦合机制[45])可避免氧气的释放及过渡金属离子的迁移,实现O2-的可逆氧化还原反应。

图 2 富锂锰基材料的两种不同反应机理[48] (a)体相反应;(b)界面反应Fig. 2 Two different mechanisms of Li- and Mn-rich cathode material[48] (a)bulk particles reactions;(b)surface reactions
同时,在富锂锰基正极材料中,晶格氧也以不可逆氧化反应的方式参与电 化学反应,并最终以氧气形式释放[46]。目前,对于富锂锰基正极材料在4.5 V处的平台,研究者广泛认同的机制是随着充电过程的进行,Li+从材料的Li2MnO3组分中持续脱出,伴随Li2MnO3中晶格氧,两者以“Li2O”的形式从材料中脱出形成空位,导致过渡金属离子迁移及Li+的不完全回嵌,材料结构遭到破坏。
因此,富锂锰基电极充放电过程,同时存在着可逆、不可逆的阴离子氧化还原反应。不可逆阴离子氧化反应会导致晶格氧析出,造成过渡金属离子迁移、结构重排(图2(a))及界面副反应(图2(b))的发生,最终造成电极不可逆容量损失,倍率及循环性能恶化[47-48]。采用离子掺杂、表面包覆手段可分别从热力学、动力学上抑制氧的释放,达到改善电极性能的效果。
2.2 过渡金属离子迁移
大量研究表明,富锂锰基正极材料在循环过程中存在过渡金属离子迁移及相转变现象。Mohanty等[49]采用中子衍射分析Li1.2Mn0.55Ni0.15Co0.1O2材料循环前后结构变化。对比发现,26次循环后,图谱中(108)特征峰强度减弱,且在0.113 nm处出现新的(440)衍射峰。这些变化证明循环后材料中出现了类尖晶石结构。这是因为,首次循环后晶格中出现大量氧空位,而氧空位的存在使Mn发生了从过渡金属层向锂层的迁移(向热力学稳定方向转化)。多次循环后Mn从锂层四面体位进一步迁移至锂层八面体位并钉扎于此,形成一种类尖晶石的晶格结构。
基于中子衍射分析结果,Mohanty等从原子角度进一步揭示了材料由层状向尖晶石结构转变的过程(图3):(1)当电压充电至3.2 V时,锂层中Li从锂层八面体(3b位)中脱出,形成八面体空位;(2)当形成足够多的锂空位时(电压升至4.1 V),锂层中八面体位的Li迁移至邻近锂层四面体空隙,形成四面体位Li。随后,过渡金属层中八面体位的Li也迁移至锂层四面体空隙,与过程(1)形成的四面体位Li及过渡金属层中的空位形成“Li—⊙—Li”(⊙表示空位)哑铃结构[24];(3)当电压 ≥4.1 V,过渡金属层中Mn通过氧空位迁移至锂层四面体位[50];(4)电压继续升高(≥ 4.5 V),Mn从锂层四面体位迁移至锂层八面体位。至此,材料结构发生了从层状到尖晶石的转变。

图 3 富锂锰基正极材料由层状到尖晶石结构相变机理[49]Fig. 3 Proposed mechanism transforming from layered to spinel structure of Li- and Mn-rich cathode materials[49]

图 4 富锂锰基正极材料[51] (a)结构演变示意图;(b)充放电曲线Fig. 4 Li-and Mn-rich cathode materials[51] (a)schematic diagram showing the structure evolution;(b)charge-discharge curvesduring cycling
Gu等[51]认为过渡金属离子迁移导致的结构转变首先发生于材料表面,而后逐渐向内部生长,并最终扩散至颗粒体相(图4)。另外,对于材料中的及C2/m相,由层状结构向尖晶石结构的转变方式不同,导致最终形成的尖晶石晶粒具有不同结构特征(图4(a))。目前认为,层状结构向尖晶石结构的转变是富锂锰基电极容量/电压衰减的主要原因[52-53]。从图4(b)中可以看出,电极100周循环后结构发生转变,中值电压由第4周的3.57 V降至3.37 V。大量实验表明,采用离子掺杂等手段可显著提高材料结构稳定性,抑制充放电过程中的相变,进而提高电极循环稳定性。
2.3 界面副反应
2.3.1 电解液与析出氧的反应
富锂锰基正极材料/电解液界面发生副反应导致电极性能恶化,这与晶格氧的不可逆释放密切相关。Castel等[54]采用同步实验气体分析手段检测到Li[Li0.2Ni0.2Mn0.6]O2材料在首次充电至4.6 V时有O2释放,界面氧通过亲核效应催化电解液的氧化分解并在正极表面形成固体电解质界面膜(SEI,主 要 成 分 包 括ROCO2Li、(ROCO2)yM、ROLi、(RO)xMs、Li2CO3等[55]),导致了不可逆容量损失及电极表面阻抗增加。此外,副反应生成的其他产物如HF会严重腐蚀正极颗粒表面,加速Mn等金属元素的溶解,造成循环、倍率性能的恶化。
图5阐明了富锂锰基正极材料充电时界面的副反应过程:(1)充电至4.2 V,电解液中碳酸乙烯酯(EC)发生氧化分解,伴随CO2释放及EC衍生物的生成;(2)电压增大至4.5 V,氧阴离子氧化生成O2(Li2MnO3相的活化过程),界面O2催化EC及衍生物的氧化分解,伴随新的CO2及中间产物的生成;(3)电压继续升至4.7 V,中间产物进一步分解,继续生成CO2、O2及其他副产物。不可逆反应产物持续堆积,电极表面阻抗增大[54]。

图 5 富锂锰基材料界面反应机理示意图[54]Fig. 5 Surface reaction mechanisms of Li- and Mn-rich cathode materials[54]
2.3.2 过渡金属元素的溶解
在富锂锰基电极中,活性物质的损失是电压/容量衰减、库仑效率低的主要原因之一,而过渡金属元素溶解是活性物质损失的主要方式[56]。在Co、Ni、Mn等几种过渡金属元素中,Mn是最易溶解的元素[57]。其溶解机理有两种:(1)歧化反应机理。结构中Mn3+难以稳定存在,易发生歧化反应,其反应式可表示为: 2Mn3+=Mn4++Mn2+[58-59]。反应产生的Mn2+快速溶出到电解液中,造成活性物质的损失。(2)HF腐蚀机理。HF主要来源于两方面:其LiPF6反应生成HF。其反应式可表示为:2LiMn2O4+4H+=2Li++3MnO2+Mn2++2H2O[60]。Mn的 溶 解造成活性物质损失,正极颗粒表面发生相变[61],同一,电解液中的微量水分与LiPF6反应生成HF。其二,高电压下电解液氧化分解产生的H+与时溶解的Mn以副产物形式在正极表面沉积,造成大的界面阻抗,降低正极的循环稳定性[62]。
综上,高电压(> 4.5 V)下,电解液易发生氧化分解,同时电极首次活化析出的氧会催化电解液的分解,使电极界面副反应更加复杂。因此,同传统层状材料比,富锂锰基电极界面稳定性更差。采用表面包覆等手段,可阻止正极颗粒与电解液的直接接触,进而抑制界面副反应的发生,改善电极性能。
2.4 小结
晶格氧析出、过渡金属离子迁移、界面副反应是材料的主要失效机制,导致了富锂锰基电极低的首次库仑效率、倍率及循环性能,限制了其商业化应用。虽然目前材料的失效机制尚未完全明了,仍需进一步研究,但面对上述挑战,研究者们发展了多种改性及制备方法,包括表面包覆、离子掺杂、结构调控生长等,有效提高了材料的综合电化学性能。表2对富锂锰基电极存在的问题、原因及对应解决策略进行了归纳总结[63-75]。

表 2 改善富锂锰基电极性能的主要策略及方法Table 2 Main strategies and solutions to improve the performance of Li- and Mn-rich cathode materials
3 改性方法
3.1 离子掺杂
离子掺杂指选择与所替换对象半径相近的离子进行掺杂,以改善材料导电性;形成更强M—O键,稳定晶体结构;增大晶胞参数,提高Li+脱嵌动力学,进而提高材料循环及倍率性[76-78]。近年来,研究者通过离子掺杂方法,使富锂材料性能得到大幅提高(表3)。如Qing等[79]采用NaCl高温熔融盐浸渍法对富锂锰基正极材料进行表面Na+梯度掺杂。掺杂进的Na+通过钉扎效应使锂层结构更加稳定,同时提高了材料导电率,使电极具有286 mAh/g的放电比容量及87%的首周库仑效率。Li等[66]选用K+对材料进行掺杂改性,结果表明,该法可提高Li1.2Mn0.54Co0.13Ni0.13O2材料结构稳定性,抑制充放电过程层状结构向尖晶石结构的转变。Wang等[68]运用喷雾热解法成功将Mg2+掺入Li1.2Ni0.13Co0.13Mn0.54O2富锂材料,结果表明,Mg2+的存在有效抑制了过渡金属离子向锂层的迁移,缓解了过渡金属离子对Li+扩散的阻碍。此外,Shang等[80]利用稀有金属元素Ru对Li1.2Ni0.13Co0.13Mn0.54O2进行掺杂后发现材料循环、倍率性能均得到提高。通过分析认为,这主要是由于Ru4+/5+的4d轨道与O2-的2p轨道重叠较小,增强了金属原子M与O间的键能,因此可抑制高电压下分子氧的释出,使材料各项性能同步提高。

表 3 离子掺杂对富锂锰基正极材料的改性效果Table 3 Effect of ions doping on electrochemical performance of Li- and Mn-rich cathode materials
除了用阳离子对富锂锰基正极材料进行掺杂改性外,也有研究者尝试了用阴离子进行掺杂。如Li等[81]采用共沉淀法制备出F-掺杂的富锂锰基正极材料,结果表明,F-的引入使材料具有优异循环特性,这是因为F-取代了部分O2-,使材料首周循环释放的氧减小,有效抑制了材料由层状向尖晶石结构的转化,起到了稳定晶体结构的作用。同时掺杂后的材料晶面间距增大,利于Li+的传输,倍率性能也由此提高。Li等[82]利用硼氧聚阴离子BO33-、BO45-对Li[Li0.2Ni0.13Co0.13Mn0.54]O2材料进行共掺杂以调控其电子结构。结果表明,掺杂制备的Li[Li1.2Ni0.13Co0.13Mn0.54](BO4)0.015(BO3)0.005O1.925具有更强的M—O键及更低的O2p轨道能位,从而有效抑制了氧的释放,使材料具备优异的循环及热稳定性。另外,Chen等[83]对富锂锰基正极材料进行Cd2+和S2-共掺杂,材料100周循环的电压降为0.25 V,较未改性材料,电压提升了0.15 V。
3.2 表面包覆
表面包覆能有效保护电极材料,抑制正极颗粒与电解液的界面副反应,同时能够在一定程度上阻挡氧的释出,提高可逆容量,改善循环性能[89-90]。按作用机理,包覆层可分为惰性包覆层(如MgO、SnO2、CeO2、AlF3、CoF2、MgF2、CePO4)、电子电导包覆层(如聚乙烯二氧噻吩)、离子电导包覆层(如Li2ZrO3、Li4Ti5O12、LiCoPO4)及活性包覆层(如MnO2、Nb2O5、Co3O4)。表4列举了不同材料包覆改性的实例。其中,Shi等[91]通过熔盐浸渍法在Li[Li0.2Mn0.54Ni0.13Co0.13]O2表面包覆了MgO,该惰性保护层阻止了电解液与材料的直接接触,抑制了两者界面间的副反应,使材料展现出优异循环性能。Ju等[74]用聚乙撑二氧噻吩(PEDOT)-聚乙二醇(PEG)双导电聚合物包覆LiNi0.6Co0.2Mn0.2O2材料,改性后材料导电性得到大幅提高,电极容量、循环性能均得到提升。Jin等[92]在富锂锰基正极材料表面包覆了β-MnO2材料,其拥有的储锂特性允许放电过程更多的Li+回嵌,减小不可逆容量损失。改性后的材料首次库仑效率由61.2%提高至88.4%。此外,Pan等[93]将Nb2O5作为包覆层改性富锂锰基正极材料,材料倍率及循环性能得到同步提高。这是由于Nb2O5不仅可阻止正极颗粒与电解液的直接接触,本身还具有良好导离子特性,提高了Li+扩散动力学。
另外,不少研究者从双相、多相包覆角度进行研究,协同发挥多种材料的优势,以提高材料综合性能。Chen等[94]合成了Li1.2Ni0.13Co0.13Mn0.53O2@AlF3@石墨烯复合材料,其中AlF3惰性保护层的存在抑制了过渡金属元素的溶出,稳定了材料结构;而外层包覆的石墨烯提供了长程导电网络,保证了电极具有良好电子导电性,从而使电极展现优异倍率及循环性能。

表 4 表面包覆对富锂锰基正极材料的改性效果Table 4 Effect of surface coating on the electrochemical performance of Li- and Mn-rich cathode materials
3.3 晶体结构调控
3.3.1 异质结构设计
近年来,设计异质结构的富锂锰基正极材料成为研究热点。Wang等[105]通过控制过渡金属元素含量来抑制材料的电压衰减现象。制备的Li1.14[Mn0.6Ni0.25Co0.15]0.86O2微球,从表层到球心,Mn元素的含量逐渐增加,而Co元素的含量逐渐减少。这种成分梯度设计抑制了材料由层状向尖晶石结构的转变,改善了电压衰减情况,从而使电极表现出良好的循环性能。Luo等[71]通过溶剂热法制备出Li1.2Mn0.4Co0.4O2材料,结合XRD、HRTEM等分析表征,不同焙烧温度下制备的材料具有不同结构,其中700 ℃下焙烧的样品含有尖晶石结构(图6(a)),而800 ℃焙烧样品无该结构特征(图6(b))。形成的尖晶石结构可为Li+的快速扩散提供三维通道,因而700 ℃样品电极倍率性能优异,1200 mA/g电流下,放电比容量可达185 mAh/g。Yang等[106]报道了具有核壳结构的Li1.15[(Mn1/3Ni1/3Co1/3)0.5(Ni1/4Mn3/4)0.5]0.85O2材料,该异质结构材料与核成分Li1.15(Mn1/3Ni1/3Co1/3)0.85O2相比具有更优异循环性,与壳成分Li1.15(Ni1/4Mn3/4)0.85O2相比则展现出更优异倍率特性。此外,Xu等[107]通过控制混锂量,合成了具有异质结构的Li1.26-xNi0.11Co0.04Mn0.59O2,该微米球材料展现了优异电化学性能,0.2 C倍率下循环100周,放电比容量达286 mAh/g。

图 6 Li+扩散示意图[71] (a)700 ℃煅烧样品;(b)800 ℃煅烧样品Fig. 6 Schematic illustration for Li+ diffusions within the microspheres prepared after calcination[71] (a)700 ℃;(b)800 ℃
3.3.2 多级微纳结构设计
多级微纳结构设计是提高富锂锰基正极材料综合性能的一种有效途径,可兼得纳米、微米材料的优势特点:不仅具备纳米材料本征载流子扩散路径短的优点,也具有微米材料表面能低、不易团聚、化学稳定性高等特征。因此,设计合成多级微纳结构可望同时解决富锂锰基正极材料普遍存在的倍率性能不佳、稳定性差等问题。Jiang等[108]以多孔MnO2微米球为模板,制备出0.3Li2MnO30.7LiNi0.5Mn0.5O2中空微米球,同一般固相法及溶胶-凝胶法制备的材料相比,具有更优异的倍率及循环性能。Zhang等[109]利用水热法设计制备出Li1.2Ni0.13Mn0.54Co0.13O2多级结构材料,材料由零维纳米颗粒自组装形成微米多孔棒状结构,独特的结构特征使材料表现出优异的循环、倍率及热稳定性。Chen等[110]采用聚乙二醇辅助共沉淀法,制备出0.5Li2MnO30.5LiMn1/3Ni1/3Co1/3O2材料,独特的多孔结构既可保证电解液的充分浸润,又可缓冲充放电过程活性材料的体积应变,从而使材料表现出较高的倍率及循环性能。
3.3.3 晶面调控生长
晶面调控生长也是改善富锂锰基正极材料电化学性能的有效方法,近几年有不少学者对此展开了研究。如Wei等[75]通过控制前驱体合成工艺及固相烧结制度,设计制备出具有(010)晶面取向的片状Li(Li0.17Ni0.25Mn0.58)O2材料。如图7所示,晶体沿着垂直某晶向方向生长,而平行于该方向的晶面在生长中趋于消失。结合第一性原理计算,活性晶面(010)有利于扩展Li+在材料体相中的传输通道,极大提高Li+的脱嵌动力学。电极在6 C倍率下放电,比容量达200 mAh/g,展现了突出的倍率特性。而Luo等[111]通过共沉淀法分别制备出沿着[101]及[001]方向生长的富锂锰基正极材料。结果表明,具有(101)晶面取向的材料具有优异倍率特性,而具有(001)晶面取向的材料在抑制电压/容量衰减方面更具优势。此外,Chen等[112]通过共沉淀法制备出由一次纳米片组装而成的二次微米球,材料具有的(010)取向特征显著提升了充放电过程Li+的扩散速度。因此,最终制备的电极倍率特性优异,20 C倍率下,放电容量可达141.7 mAh/g。
3.4 其他方法

图 7 具有{010}及{001}晶面取向的Li(Li0.17Ni0.25Mn0.58)O2材料生长示意图[75]Fig. 7 Schematic illustration for growth of Li(Li0.17Ni0.25Mn0.58)O2 with exposed {010} and {001}planes[75]
电极中活性材料表面具有高电化学活性,因此,表面改性是提高富锂锰基正极材料性能的有效方式。除上述提到的表面包覆研究,其他的表面改性工作,如贫Ni[113]、氧空位[114]、NiCo量子点修饰[115]等也获得一定进展。美国阿贡实验室Kang等[69]采用稀硝酸处理富锂锰基正极材料0.5Li2MnO30.5 LiCo0.25Ni0.44Mn0.31O2(实质为预先活化结构中Li2MnO3组分),研究发现,随着处理时间的增加,材料首周充电比容量逐渐下降,而放电比容量基本保持不变,最终电极首周库仑效率可达100%。Zheng等[116]采用一定浓度的Na2S2O8溶液处理Li[Li0.2Mn0.54Ni0.13Co0.13]O2,使Li、O从材料中预先脱出形成空位,同时在材料表面形成尖晶石结构包覆层。改性后材料首效达100%,且1 C放电倍率下比容量达200 mAh/g。Li等[117]将Li1.2Mn0.6Ni0.2O2与NH4HCO3按一定化学计量比混合,Ar气氛下600 ℃煅烧10 h,制得含有氧空位的富锂锰基正极材料。研究表明,氧空位的构筑提高了M—O间键能,使O2p轨道能量降低,有效抑制了氧的不可逆释放,同时诱导产生尖晶石相,为Li+扩散提供三维通道。经改性,材料首次比容量由216.1 mAh/g提高至316.3 mAh/g,库仑效率由80%提升至94.8%。此外,Zhang等[118]和Erickson等[119]将富锂锰基正极材料在氨气气氛中进行400 ℃渗氮处理,所获材料的首效、倍率、循环性能均得到明显提升。
另外,也有不少研究者对电解液、黏结剂的优化做了深入研究。如Han等[64]通过在电解液中添加功能化添加剂TMSP(trimethylsilyl phosphite),在Li1.17Ni0.17Mn0.5Co0.17O2表面形成一层薄且均匀的保护层,有效抑制了过渡金属元素的溶出及结构的不可逆相变。此外,保护层利于高倍率下Li+的扩散,从而使电极具有高的循环及倍率性能。Nayak等[120]研究发现,在EC-DMC/LiPF6体系电解液中添加4%(质量分数)的LiBOB,能显著提高Li1.2Mn0.56Ni0.16Co0.08O2材料的稳定性,50周循环后,容量保持率可达97%。而Wu等[121]采用PAN(polyacrylonitrile)及PVDF(polyvinylidene fluoride)复合物作为黏结剂,由于PAN在高电压下稳定且抗膨胀性强,同PVDF复合用于富锂锰基电极中,可显著改善电化学性能。Zhang等[122]首次将GG(guar gum)作为Li1.14Ni0.18Mn0.62O2电极黏结剂,通过对正极材料颗粒表面的紧密包覆,可有效抑制正极颗粒/电解液的界面副反应,同采用常规黏结剂PVDF比,显著改善了容量/电压衰减情况。此外,新型黏结剂羧基纤维素钠[123]、海藻酸钠[124]等也能显著提高富锂锰基正极的电压稳定性。
3.5 小结
采用上述技术手段,可显著改善富锂锰基正极材料的电化学性能,但目前大部分研究仍处于实验室阶段,报道的工艺方法普遍复杂,需开发低成本、高效的改性制备技术,以实现材料的大规模商业化制备。此外,单一的改性方法具有局限性,联合改性机制将是未来富锂锰基正极材料的改性方向。另外,同商用正极材料比,富锂锰基正极材料还存在其他应用问题,如LiCoO2压实密度为4.0~4.2 g/cm3,而富锂锰基正极材料压实密度低于3.0 g/cm3,低的体积比能量限制了其的应用。
4 结束语
随着社会发展,电动汽车、消费类(3C)电子产品、储能装置等对锂离子电池的能量密度提出了更高要求。富锂锰基正极材料凭借高比容量(≈ 250 mAh/g)、高工作电压(≈ 3.6 V)及低成本优势,有望成为下一代高比能商用锂电池的正极材料。Li2MnO3相的存在是富锂锰基正极材料具有高容量的根本原因,而其活化过程也引发了一系列问题。首次库仑效率低、倍率性能差、容量/电压衰减快等问题限制了富锂锰基正极材料的工程化应用。晶格氧析出、过渡金属离子迁移、界面副反应是材料的主要失效机制。采用离子掺杂、表面包覆、晶体结构调控等技术手段,可显著改善富锂锰基材料的电化学性能。
尽管近年来国内外在富锂锰基正极材料研究方面取得了重要进展,但实现其真正的商业化应用,还面临几方面挑战:(1)材料结构、电化学反应机理及失效机制等尚未完全明了,需利用先进表征手段揭示材料合成及循环中的结构演变,材料的微观结构及电化学性能间的构效关系有待更深入、系统研究;(2)目前大部分研究仍处于实验室阶段,报道的工艺方法普遍复杂,需开发低成本、高效的改性制备技术,实现材料的大规模商业化制备;(3)与富锂锰基材料相匹配的高电压电解液、黏结剂等的研究工作也需全面开展。单一的改性方法具有局限性,联合改性机制将是未来富锂锰基正极材料的改性方向;(4)应满足电池行业对正极材料的其他要求,如富锂锰基正极材料的振实密度仍需提高。
猜你喜欢
起重运输机械(2023年22期)2023-12-12 09:55:52
安徽电子信息职业技术学院学报(2019年2期)2019-04-26 06:38:28
汽车电器(2018年1期)2018-06-05 01:23:04
制造业自动化(2017年2期)2017-03-20 14:26:08
电源技术(2016年2期)2016-02-27 09:04:42
中国塑料(2015年6期)2015-11-13 03:03:05
电源技术(2015年12期)2015-08-21 08:58:54
电源技术(2015年12期)2015-08-21 08:58:20
风能(2015年8期)2015-02-27 10:15:12
风能(2015年5期)2015-02-27 10:14:46
