
乳粉中维生素K1含量不确定度评定分析
2021-02-15 07:42罗渺琴
食品安全导刊 2021年30期
罗渺琴
(广东东泰乳业有限公司公司,广东揭阳 515500)
高效液相色谱法、气相色谱-质谱法、液相色谱-串联质谱法均属于乳粉中维生素K1含量测定的常用方法,在大批量检测方面,本文研究的高效液相色谱法表现突出,其他两种方法测定成本较高,不适合开展大批量检测。但如果乳粉的基质较为复杂,高效液相色谱法可能无法满足检测需求,为保证高效液相色谱法更好服务于相关检测,本文围绕乳粉中维生素K1含量不确定度评定开展具体研究。
1 材料与方法
1.1 材料与试剂
氢氧化钾、脂肪酶、氯化锌、冰乙酸、正己烷、碳酸钾、无水乙酸钠、甲醇、四氢呋喃、无水乙醇和维生素K1标准品,其中甲醇、四氢呋喃均为色谱纯,无水乙酸钠、磷酸二氢钾分别为优级纯、分析纯,维生素K1标准品拥有99%以上纯度[1]。
1.2 仪器与设备
高效液相色谱仪、反相色谱柱,型号分别为U-3000、AQC18,反相色谱柱(250 mm×4.6 mm,5 μm)。
1.3 实验方法
1.3.1 样品前处理
按照0.000 1 g精度称取乳粉样品2.0 g置入具塞三角瓶(150 mL),加入纯水7 mL,温度控制在50 ℃,摇匀使乳粉样品充分溶解,之后将磷酸盐缓冲液加入其中并混匀,加入量为5 mL。加入脂肪酶0.2 g,充分混合后在恒温水浴振荡器中进行2 h振荡处理,温度设置为(37±2)℃,确保充分酶解。
试样酶解好后分别加入碳酸钾1 g及无水乙醇10 mL,混合均匀后将水、正己烷各10 mL加入其中,振荡或涡旋提取10 min,离心5 min,速度设置为6 000 r/min,转移上清液至旋转蒸发瓶(100 mL)中,之后加入正己烷10 mL,重复操作,进一步向旋转蒸发瓶中转移上清液。旋蒸至近干,氮气吹干后加入甲醇5 mL并摇匀,通过0.22 μm滤膜进行过滤,滤液待进样
1.3.2 色谱条件
参考《食品安全国家标准 食品中维生素K1的测定》(GB 5009.158—2016)内容,设定流速为1 mL/min,激发波长、发射波长分别为243 nm、430 nm,进样量为10 μL。实验用数学模型为:

式中:V1-提取液总体积,mL;V3-定容液总体积,mL;V2-分取的提取液体积,mL;X-维生素K1含量,μg/100 g;ρ-试样溶液中维生素K1浓度,ng/mL;m-试样质量,g。
不确定度的来源可细分为6个方面,包括样品质量引入、标准品校正引入、样品体积引入、仪器引入、标准曲线引入、样品测量重复性引入的标准不确定度[2]。
2 维生素K1含量不确定度评定
2.1 样品质量引入
实验过程需要称取样品2.000 0 g,由于存在0.5 mg的扩展不确定度及具体值为2的包含因子,可确定其存在1.25×10-3相对标准不确定度,计算过程为:

2.2 标准品校正引入
2.2.1 标准工作液引入的合成相对不确定度
(1)不确定度会因标准溶液校正引入。容量瓶(100 mL)定容标准工作液存在±0.10的最大允许误差,属于三角分布,因此可确定容量瓶存在4.08×10-4的相对标准不确定度,计算过程为:

(2)移液管(1 mL分度)在配置标准工作液过程中应用,其存在0.008的最大允许误差,属于三角分布,因此可确定存在3.27×10-3相对标准不确定度,计算过程为:

对于20 ℃条件下校正的玻璃器具,结合(20±5) ℃的实验室温度,可通过估算体积膨胀系数和温度范围确定温度波动带来的不确定度,同时可忽略远小于液体膨胀系数的玻璃膨胀系数。
(3)通过甲醇稀释进行标准工作液配制,移取使用移液管(1 mL分度)并在容量瓶(100 mL)中定容,基于20 ℃条件下0.001 2(1/℃)的甲醇膨胀系数,标准不确定度计算需假设存在矩形分布的温度变化,因此可得到3.46×10-3标准不确定度,计算过程为:
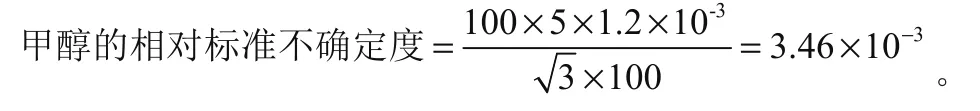
基于标准工作液进行分析,用urel(Ca)、urel(Cb)、urel(Cc)分别代表容量瓶、移液管、甲醇引入的相对标准不确定度。最终可得到5.90×10-3的合成相对标准不确定度,计算过程为:

2.2.2 标准品校正时的标准不确定度
还需要围绕仪器在标准品校正时的标准不确定度进行分析,结合仪器存在的0.5%扩展不确定度及具体值为2的包含因子[3],可确定相对标准不确定度为2.5×10-3,计算过程为:

因此存在6.41×10-3的标准品引入合成相对标准不确定度,用urel(C0)、urel(C1)分别代表标准工作液、分光光度计校正引入的合成相对标准不确定度。计算过程为:

2.3 样品体积引入
旋转蒸发仪蒸干的样品需要对剩余样品进行甲醇溶解处理,这一过程使用移液管(5 mL分度),其存在±0.025的最大允许误差,属于三角分布,可由此确定其存在2.04×10-3的相对标准不确定度,计算过程为:

采用相同方法,基于20 ℃条件下0.001 2(1/℃)的甲醇膨胀系数且假设存在矩形分布的温度变化,温度波动存在3.47×10-3相对标准不确定度。围绕样品体积进行分析可以确定,其存在4.03×10-3引入的合成相对标准不确定度[4]。
2.4 仪器引入
结合检定证书信息可以确定,实验使用的高效液相色谱仪仪器存在0.1%的相对扩展不确定度及具体值为2的包含因子,因此可确定存在5×10-4相对标准不确定度,计算过程为:

2.5 标准曲线引入
开展0.380 μg/mL、0.190 μg/mL、0.095 μg/mL、0.038 μg/mL、0.019 μg/mL和0.01 μg/mL质量浓度的维生素K1标准工作曲线配制,分别开展1次测量,标准溶液质量浓度-峰面积曲线通过最小二乘法拟合,标准曲线为[5]:
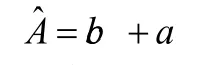
式中:a、b、c、分别为标准溶液的浓度(校准用)、斜率、截距、峰面积,ˆ与校准用标准溶液浓度对应。
结合计算获得的数据分析截距、斜率、相关系数关系可以发现,截距、斜率、相关系数的值分别为24.595 93、21 077.995 2、0.999 92,因此可得到校准曲线A,即:
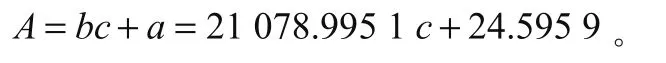
开展奶粉试样平行测定2次,可确定样1、样2的峰面积分别为5 540.22、5 563.56,浓度分别为0.261 μg/mL、0.264 μg/mL,含量分别为65.413 88 μg/100 g、65.692 82 μg/100 g,结合平均试样浓度和平均峰面积,可确定2次重复测定能够带来0.663%的相对标准不确定度,计算过程为:

2.6 样品测量重复性引入
开展维生素K1平行测试,目标为同一批次乳粉试样,总的分析数据随机变化能够由此获取,如测定次数为1、3、7、10 时的测定结果分别为 65.4 μg/100 g、66 μg/100 g、63.6 μg/100 g、60.6 μg/100 g,10 次平均值为 64.2 μg/100 g。围绕重复测量样品开展评定,平均值标准偏差用于平均值的标准不确定度表征,可得到8.03×10-3的相对标准不确定度,计算过程需结合所测结果r的平均值标准偏差及标准不确定度,分别表示为可得到计算公式:

2.7 相对标准不确定度合成
结合测量样品质量、标准品校正、样品体积、仪器、标准曲线和样品测量重复性共6种来源的相对标准不确定度,可确定乳粉中K1测定过程中高效液相色谱法的不确定度,即0.977 25 μg/100 g,用urel(C)、urel(V)、urel(m)、urel(q)、urel(s)、urel(r)分别代表标准品、样品定容体积、样品质量、标准曲线、仪器、测量重复性的相对标准不确定度。计算过程为:

2.8 扩展不确定度
结合数值为2的包含因子及95%的置信水平,可通过计算得到扩展不确定度,具体数值为1.954 5 μg/100 g,计算过程为:

3 结论
通过对实验过程进行分析可以发现,乳粉维生素K1测定中,高效液相色谱法的实用性较高,能够确定样品质量、标准品校正、样品体积、仪器、标准曲线和样品测量重复性等方面引入的不确定度,高效液相色谱法应用中受到的最小二乘法拟合曲线影响最大,因此可以确定其涉及的浓度值属于不确定度测量的主要影响因素,在高效液相色谱法的应用中必须对其进行针对性控制,以此得到可靠性更高的检测结果,同时保证测量结果间存在更高的可比性,为测定乳粉维生素K1含量提供更好的支持。
猜你喜欢
食品科学(2023年4期)2023-03-06
环境保护与循环经济(2021年7期)2021-11-02
中国乳业(2020年12期)2020-04-12
制造技术与机床(2017年9期)2017-11-27
西南石油大学学报(自然科学版)(2015年3期)2015-04-16
河北工业科技(2015年4期)2015-02-27
食品安全导刊(2014年8期)2014-10-21
食品工业科技(2014年9期)2014-03-11
电加工与模具(2014年3期)2014-02-24
中国机械工程(2011年15期)2011-05-30
