
基于药物基因组学的选择性5-羟色胺再摄取抑制剂个体化治疗抑郁症*
2021-02-04 03:01周长凯徐龙张斌纪洪艳邢晓敏郭切张莎莎王文晓邵延琳于红霞李静荆凡波
医药导报 2021年2期
周长凯,徐龙,张斌,纪洪艳,邢晓敏,郭切,张莎莎,王文晓,邵延琳,于红霞,李静,荆凡波
(青岛大学附属医院药学部,青岛 266000)
2017年世界卫生组织(WHO)数据表明,2005—2015年全球抑郁症患者数量增加18.4%,2015年全球患者已达3.22亿例。选择性5-羟色胺(5-HT)再摄取抑制剂(selective serotonin reuptake inhibitors,SSRIs)是治疗抑郁症的一线药物,但严重抑郁症患者SSRIs初始治疗失败率将近50%[1],此外,这类药物的失眠、性功能障碍不良反应发生率高,在美国每年约有25 000例患者因相关不良事件就医[2]。研究证实CYP2D6和CYP2C19基因型与SSRIs代谢、疗效具相关性,可指导SSRIs治疗以提升疗效,并减少不良事件的发生[3-4]。然而在我国,临床医师对此认识相对不足且临床实践较少,制约临床抑郁治疗水平进一步提升。笔者在本研究基于CYP2D6和CYP2C19基因型对SSRI治疗的影响进行系统综述,为广大医药同行提供参考。
1 SSRIs药物
SSRIs是当前治疗抑郁症的一线药物,我国临床使用SSRIs药物有帕罗西汀、氟伏沙明、氟西汀、西酞普兰、艾司西酞普兰、舍曲林。该类药物能有效降低突触前5-HT再摄取泵60%~80%的活性,从而提高突触间隙5-HT浓度发挥治疗抑郁作用[5]。除帕罗西汀对胆碱能受体有轻微拮抗作用外,其他SSRIs都不会显著影响胆碱能受体、α-肾上腺素能受体或组胺受体。此外,帕罗西汀、氟伏沙明和氟西汀代谢主要受CYP2D6基因型影响,西酞普兰、艾司西酞普兰和舍曲林主要受CYP2C19影响[6-7]。
2 CYP2D6、CYP2C19基因多态性
除CYP2D6、CYP2C19外,SSRIs亦是其他代谢酶如CYP1A2、CYP2C9和 CYP3A4的底物,但其他代谢酶基于药物基因组学的个体化用药缺少有效的证据支持,故本文仅对CYP2D6、CYP2C19基因多态性作探讨。CYP2D6具有高度基因多态性,现已知存在100余种等位基因变异[8],其等位基因存在地域、种族多样性且出现频率有显著差异,等位基因功能分类包括正常功能(如CYP2D6*1和*2),功能降低(如CYP2D6*9、*10和*41) 和功能缺失(如CYP2D6*3~*6)。等位基因变异产生4种代谢型:超快代谢型(ultra-rapid metabolizer,UM)、强代谢型(extensive metabolizer,EM)、中代谢型(intermediate metabolizer,IM)和弱代谢型(poor metabolizer,PM)[9]。在人群分布上,功能缺失等位基因在高加索人中出现频率较高,最常见CYP2D6*3、*4、*5,其中*3、*4是PM突变的常见类型,而这种基因突变在非洲人和非裔美国人发生率仅有6%~7%。功能降低等位基因中亚洲人占比较高,其中CYP2D6*10在东方人群的出现频率超过50%[10]。正常功能等位基因中高加索人占71%,最常见类型为CYP2D6*2。与CYP2D6相似,CYP2C19由于在人群中等位基因分布频率的明显不同而表现出显著的基因多态性,已知30多种等位基因变异,然而大部分患者携带*1、*2或*17等位基因,*2等位基因在黑种人、黄种人和白人的分布频率分别为15%,29%,12%。CYP2C19等位基因功能分类如下:*1为正常功能;*2~*8为功能缺失或降低;*17为功能增强。CYP2D6、CYP2C19不同基因型的表型见表1,2。
3 CYP2D6、CYP2C19表型与SSRIs的剂量选择
3.1CYP2D6表型与帕罗西汀、氟伏沙明的剂量选择 帕罗西汀在体内吸收、消除较慢,其主要经CYP2D6代谢[11],该酶易发生饱和,故帕罗西汀会出现过量蓄积的现象,个体间的药动学参数有较大差异[12-14]。2017版神经精神药理学与药物精神病协会(AGNP)的精神科治疗药物监测共识指南推荐帕罗西汀谷浓度的治疗窗为20~65 ng·mL-1[15]。多项研究表明[16-19],与CYP2D6强代谢者相比,帕罗西汀在CYP2D6超强代谢者中的血浆浓度更低甚至无法检出,而这部分患者存在一定的治疗失败风险。虽然目前仍未明确界定帕罗西汀的最低治疗浓度,但血浆浓度较低是抑郁治疗失败的危险因素[20],超强代谢的抑郁患者应考虑使用一种不被CYP2D6广泛代谢的SSRI,关于CYP2D6超强代谢者的帕罗西汀最佳初始剂量目前仍缺少研究数据。同样,精神科治疗药物监测共识指南推荐氟伏沙明谷浓度的治疗窗为60~230 ng·mL-1 [21],CYP2D6超强代谢型对氟伏沙明治疗影响的研究证据有限,此类患者选择一种不被CYP2D6广泛代谢的SSRI可能是合理的。对于CYP2D6强或中代谢者,尽管可以预期中代谢者使用帕罗西汀或氟伏沙明时暴露量会增加,但现有研究并不支持对这两个药物进行剂量调整。
研究表明,当给予相同剂量的帕罗西汀或氟伏沙明时,与强代谢者比较,CYP2D6弱代谢者药物暴露显著增加[21-23],导致发生药物相关不良反应的风险升高[24-25],美国食品药品管理局(FDA)提示在CYP2D6活性降低患者中应谨慎使用氟伏沙明。对于弱代谢者,应选择使用一种不被CYP2D6广泛代谢的SSRI以预防不良反应的发生。当必须使用帕罗西汀或氟伏沙明时,应将帕罗西汀剂量减少50%、氟伏沙明剂量减少30%,但是考虑到药物剂型,将氟伏沙明剂量降低30%难以实现,故可将其剂量降低25%~50%,以预防不良事件发生[26]。基于CYP2D6表型的帕罗西汀、氟伏沙明给药建议见表3,4。
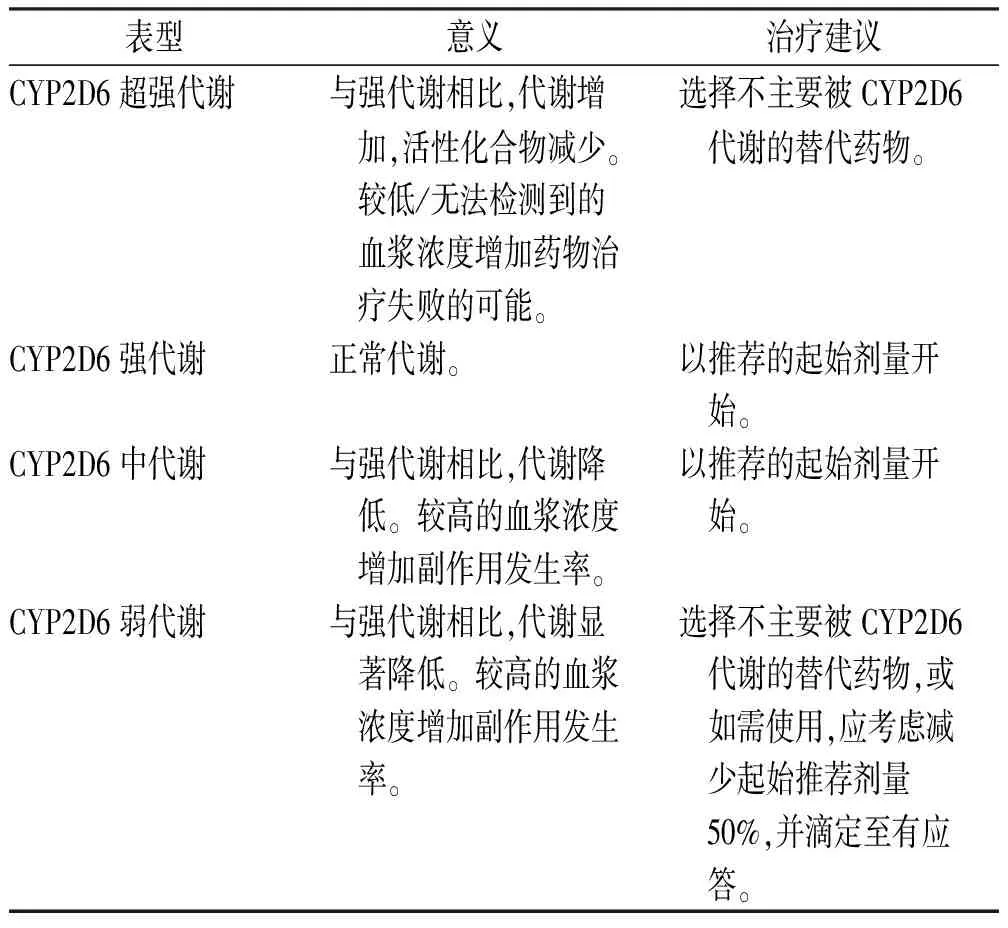
表3 基于CYP2D6表型的帕罗西汀给药建议
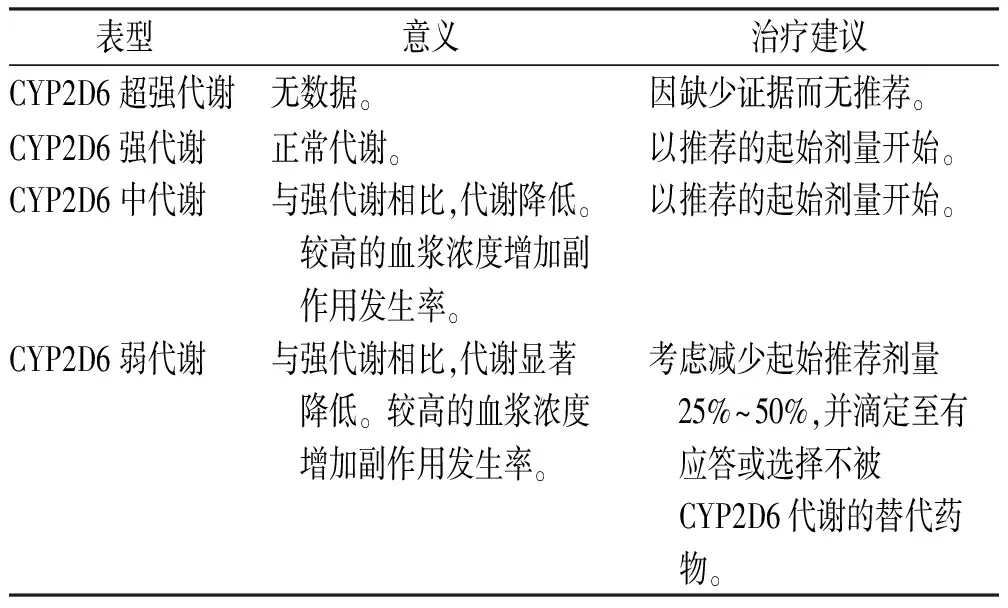
表4 基于CYP2D6表型的氟伏沙明给药建议
3.2CYP2C19表型与西酞普兰、艾司西酞普兰和舍曲林的剂量选择 艾司西酞普兰是西酞普兰的一种立体异构体,在体内主要经CYP酶系代谢,其首先通过去甲基化生成S-去甲基西酞普兰,然后进一步去甲基化产生S-双去甲基西酞普兰[27]。CYP的3种同工酶CYP2C19、CYP3A4、CYP2D6可能参与艾司西酞普兰代谢,而GUTIERREZ等[28]研究结果显示,当CYP3A4抑制剂利托那韦与艾司西酞普兰联用时,对艾司西酞普兰的代谢并无影响。HERRLIN等[29]研究表明,CYP2C19弱代谢型患者的艾司西酞普兰血药浓度明显升高,而CYP2D6弱代谢型患者无此现象,提示其主要通过CYP2C19代谢,CYP3A4与CYP2D6可能对其代谢影响较小。CYP2C19基因多态性会显著影响西酞普兰的血药浓度,是影响西酞普兰抗抑郁疗效的关键因素[30]。与强代谢者比较,CYP2C19超强代谢者中西酞普兰、艾司西酞普兰的暴露量显著降低,从而导致治疗失败[31-34]。由于目前仍缺少足够的研究数据来计算CYP2C19超强代谢者西酞普兰、艾司西酞普兰的初始剂量,若并用药物及其他临床因素允许,应选择一种不被CYP2C19广泛代谢的SSRI。CYP2C19*17纯合子具有比CYP2C19*17杂合子更强的代谢能力,可能从替代疗法中有更大获益[32-33]。
尽管CYP2C19中代谢者的西酞普兰、艾司西酞普兰血浆浓度可能升高,但现有研究并不支持对CYP2C19中代谢、强代谢者进行剂量调整,而在弱代谢者中这两种药物血浆浓度会升高,增加不良反应的发生风险[35-38],应选择使用一种不被CYP2C19广泛代谢的SSRI以预防潜在的药物不良事件。当弱代谢者必须使用西酞普兰或艾司西酞普兰时,应考虑将初始剂量降低50%[26]。对于西酞普兰,为避免QT间期延长的风险[39],FDA建议将弱代谢者的剂量降低50%,且成人最大剂量为20 mg·d-1[40]。
CYP3A4与CYP2D6不参与舍曲林的N-去甲基化代谢,但CYP2C19与它具有较高亲和力,并直接参与其代谢。此外,不同的CYP2C19基因型导致舍曲林药动学参数有显著差异,药动学数据显示在CYP2C19弱代谢者中舍曲林的口服清除率降低,而在超强代谢者中代谢仅略微增加[41-42]。不仅如此,弱代谢患者较强代谢者有更长的半衰期和较高的血药浓度-时间曲线下面积(AUC),口服舍曲林后血浆清除率显著低于强代谢者,去甲舍曲林的达峰时间较强代谢者显著延长。强代谢者对舍曲林有一清晰的消除相,而弱代谢型患者体内并不明显[43]。以上研究提示CYP2C19基因多态性是造成舍曲林与去甲舍曲林稳态血浆水平个体差异的主要因素。同时,中代谢患者的AUC高于强代谢者,但两种基因代谢型差异并不影响舍曲林的治疗效果[44]。然而,CYP2C19弱代谢者较正常代谢者的药物副作用发生更频繁[45],因此,CYP2C19弱代谢者应将剂量减少50%,或选择一种不被CYP2C19广泛代谢的SSRI。临床药物基因组学实施联盟(CPIC)不建议对CYP2C19超强代谢者调整剂量,但是当患者对足量的舍曲林维持剂量没有应答时应考虑选择一种不主要由CYP2C19代谢的SSRI替代舍曲林。基于CYP2C19表型的西酞普兰、艾司西酞普兰和舍曲林给药建议见表5,6。
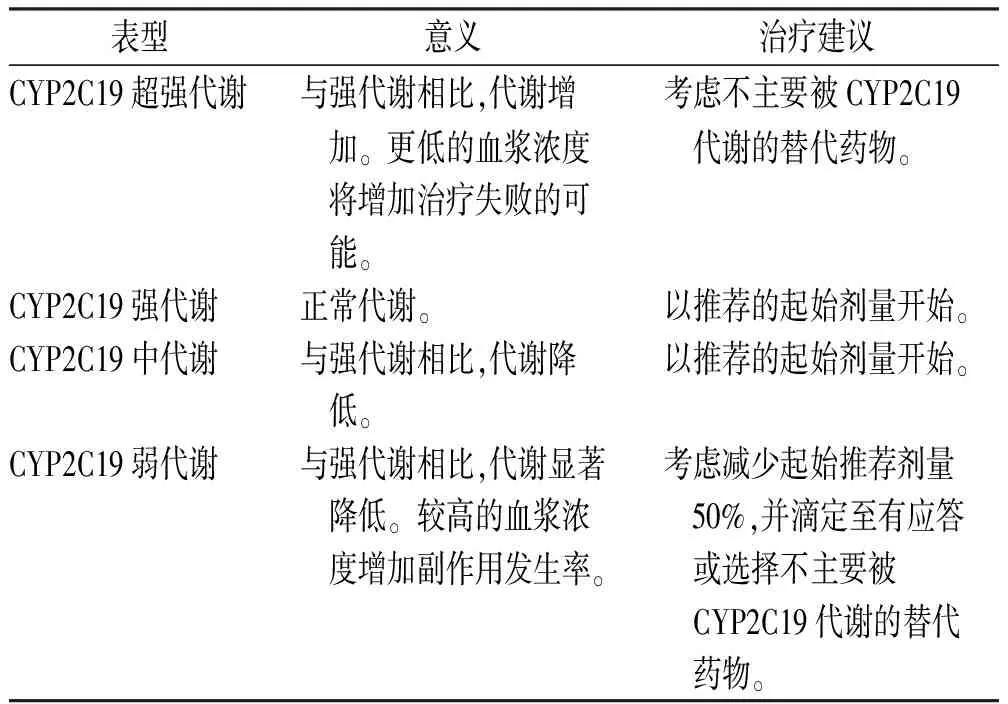
表5 基于CYP2C19表型的西酞普兰、艾司西酞普兰给药建议
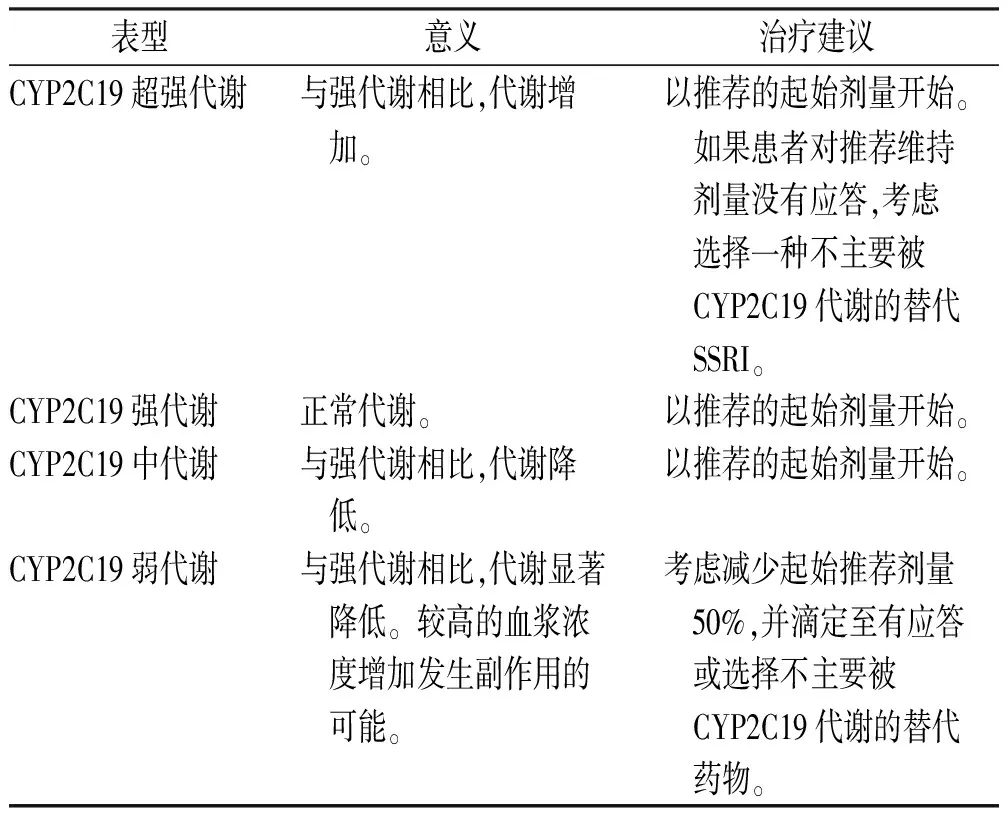
表6 基于CYP2C19表型的舍曲林给药建议
3.3CYP2D6表型与氟西汀的剂量选择 氟西汀主要经CYP酶氧化代谢,其中主要代谢途径N-去甲基代谢,其次为O-脱烷基代谢[46],去甲氟西汀为氟西汀的N-去甲基代谢物,与母体药物活性相似,半衰期更长。HAMELIN与FJORDSIDE等研究提示CYP2D6为N-去甲基代谢的主要代谢酶,其基因多态性影响氟西汀代谢[47-48]。此外,氟西汀和去甲氟西汀均为CYP2D6的强抑制剂,因代谢产物去甲氟西汀不可逆地与CYP2D6结合致其失活,故氟西汀可抑制自身代谢[49]。但当前有关CYP酶对其代谢调节的研究较少,且研究结果仍有争议。氟西汀经CYP2D6转化为S-去甲氟西汀,经CYP2D6和CYP2C9转化为R-去甲氟西汀,尽管R-去甲氟西汀的药理活性较弱,但氟西汀和R/S-去甲氟西汀均会调节5-HT的再摄取。精神科治疗药物监测共识指南推荐氟西汀谷浓度治疗窗为60~230 ng·mL-1,氟西汀和去甲氟西汀谷总浓度治疗窗为120~500 ng·mL-1。CYP2D6弱代谢者比强代谢者的氟西汀血浆浓度更高,但氟西汀加上去甲氟西汀血浆浓度的总和可能不会因CYP2D6表型不同而有显著差异。现阶段,CYP2D6表型对氟西汀和去甲氟西汀血浆浓度总和的影响,以及二者浓度间的不平衡是否影响患者预后或安全性的相关研究匮乏,故当CYP2D6超强代谢或弱代谢者使用氟西汀时需进行密切监护或考虑选择一种不被 CYP2D6广泛代谢的SSRI替代氟西汀。
4 结束语
在全球范围内,抑郁症发病率呈逐年上升之势[50-51],SSRIs作为一线治疗药物,其临床应用仍被药物疗效差、安全性低所困扰,探索基于基因多态性的SSRIs个体化治疗是进一步提升抗抑郁药物治疗水平的重要手段。我国CYP2D6、CYP2C19基因多态性理论与临床研究不断深入,以及CPIC、荷兰药物基因组学工作组等专业国际学术机构基于基因组学的SSRIs使用相关指南的发布,都为临床抗抑郁药物治疗水平的提升、抑郁患者的个体化治疗提供了参考。
猜你喜欢
中国药学药品知识仓库(2022年7期)2022-05-10
中国现代医生(2022年6期)2022-04-23
西藏艺术研究(2021年1期)2021-06-02
心理与健康(2019年11期)2019-11-15
心脑血管病防治(2016年6期)2017-01-16
中国民族民间医药·上半月(2016年12期)2017-01-11
中国实用医药(2016年23期)2016-12-26
中国实用医药(2016年27期)2016-11-30
环球时报(2016-11-10)2016-11-10
中国实用医药(2016年21期)2016-08-19
