
SLC16A2 基因变异一例报道并文献复习
2021-01-27 08:26:34邹品芳黄彦臻黄中燕刘圆圆向俊璐冉莉莉周文智
中华脑科疾病与康复杂志(电子版) 2020年3期
邹品芳 黄彦臻 黄中燕 刘圆圆 向俊璐 冉莉莉 周文智
SLC16A2 基因相关疾病称为Allan-Herndon-Dudley 综合征(Allan-Herndon-Dudley syndrome,AHDS),是一种X 连锁隐性遗传病,由编码单羧酸甲状腺激素转运蛋白8(monocarboxylate transporter 8,MCT8)SLCl6A2 基因突变所致[1-2]。临床上以严重认知运动语言发育迟缓、由婴幼儿期肌张力低下逐渐演变为痉挛性截瘫、血清甲状腺激素异常、髓鞘发育延迟为主要临床特点,可伴小头畸形、面容异常、构音障碍、脊柱侧弯、关节挛缩、反射亢进、共济失调、阵发性运动障碍、易咳呛、反复呼吸道感染等表现[3]。目前国内外AHDS 的流行病学报道罕见,发病率尚不详,本文结合相关文献就成都市妇女儿童中心医院儿童康复科于2019 年11 月收治的1 例SLC16A2 基因变异患儿的临床特征及康复治疗进行报道。
病例资料患儿男,11 月龄,因“发现竖头不稳5+月”就诊于我科门诊,家长要求入院治疗。入院前于当地行康复治疗2+月后仍发育落后,表现为竖头不稳,翻身、独坐、爬行不能,双手无主动抓握,呼名无反应及应答,偶发单元音,认知、运动、语言均全面落后。患儿系G2P2,孕40 周顺产,出生体质量3350 g,根据家长提供患儿“生后2 d 有颜面部、口唇发青吸氧治疗病史”以及“黄疸3 d 自然消退”;父母体健、非近亲结婚,G1P1 姐姐4+岁,1+岁走路、1+岁说话,就读幼儿园无特殊,否认家族类似患者及遗传代谢性疾病史。入院体检:生命体征平稳,精神可,面色红润,皮肤无黄染,无特殊面容,前囟未闭(1.0 cm×0.5 cm),平软,张力不高,无眼震,心音有力、律齐,闻及杂音,肺部、腹部查体未见明显异常。四肢肌张力降低,四肢被动活动时紧张;共济运动不能配合,肱二头肌反射、膝腱反射、跟腱反射均减弱;足握持反射、侧弯反射及病理征阳性。入院后头颅MRI 提示脑干区斑片状异常信号,心脏彩超示卵圆孔未闭,视听觉诱发电位及动态脑电图正常。格赛尔发育量表评估儿童发育,显示大运动极重度发育落后,适应性、精细运动、语言、个人-社交均重度发育落后。心肌标志物:超敏肌钙蛋白I<0.006 ng/mL、肌红蛋白<3.00 ng/mL。血脂:高密度脂蛋白胆固醇0.99 mmol/L。肝功:谷氨酰转肽酶8.4 U/L、球蛋白17.6 G/L、α-羟丁酸脱氢酶197.8 U/L。维生素B12 1255 pg/mL。首次甲状腺功能五项指标显示三碘甲状素原氨酸(total triiodothyronine,TT3)3.21 ng/mL(正常范围0.60~2.62 ng/mL)、游离三碘甲状原氨酸(free triiodothyronine,FT3)6.02 pg/mL (正常范围1.92~4.92 pg/mL)、总甲状腺素(total thyroxine,TT4)和游离甲状腺素(free thyroxine,FT4)正常、促甲状腺刺激激素(thyroid-stimulating hormone,TSH)正常;未治疗干预情况下约10 d 后复查TT3、FT3 正常,FT4 0.68 ng/dL(参考范围1.02~1.73 ng/dL),TT4、TSH 正常。凝血、电解质、同型半胱氨酸、游离脂肪酸、乳酸、肾功能、血糖、血氨、血常规、维生素D、叶酸、血液细胞形态等未见明显异常。经患儿家长知情同意后,对患儿及父母进行高精度临床外显PLUS 检测,检测到SLC16A2 基因半合变异c.467_469delTCT(p.F156del),测序数据显示这个变异遗传自患儿母亲(杂合状态),父亲无突变。OMIM 编号:300095。
该患儿入院后经康复评估,制定个体化的具体康复训练方法,治疗项目以运动疗法为主,采用Bobath 法及上田法以手法改善肌张力、抑制异常姿势反射、异常运动模式,促进直立及平衡,加强头控、诱导翻身及四肢本体感觉刺激。作业治疗以改善上肢关节活动范围、增强肌力,促进视听觉刺激及主动抓握能力。另外,根据中医辨证采用头针治疗,主穴选取运动区及足运感区为主,配合智九针加减促进运动及认知的改善;中医推拿以头项部通督点穴法、膀胱经循经推按法、推天柱骨、拿肩井、点按穴位等手法舒筋整复、调理脏腑功能,同时予以神经肌肉电刺激疗法,部位选取双侧腰背肌及肱三头肌以促进腰背部及双上肢运动神经及肌肉兴奋,通过以上综合康复治疗半个月,患儿运动、精细、认知等功能无明显改善。出院后电话随访或患儿未行治疗,临床表现无明显改变,后期需继续进行长期随访。
讨论1944 年,Allan 等[4]首次报道了AHDS。AHDS 是由MCT8 编码基因SLC16A2 突变所致[1-2]。多数研究者认为突变导致MCT8 转运甲状腺激素功能的缺失引起了神经系统发育损伤[5-8]。研究报道,MCT8 是T3 进入神经元的主要转运体蛋白,能促进T3 在细胞中的摄取,若MCT8 发生突变,将导致T3 不能进入大脑细胞内,从而导致甲状腺功能异常和神经系统发育损伤[9-10]。大多数AHDS 患者血清甲状腺功能中存在高T3、低T4、TSH 水平正常或略有升高的情况,也有报道仅1 项指标异常、基因确诊患者[2,11-12]。本例患儿初次甲状腺功能为高T3、正常T4 及TSH,未药物干预情况下复查甲状腺功能正常T3 及TSH、低T4,日本曾有报道仅T3 升高、基因确诊患者[13]。有研究者认为,尽管有些患者不存在甲状腺功能异常的临床体征,但甲状腺激素水平的紊乱可能成为一些可疑患者进行早期系统检测筛查的提示[5]。AHDS 是一种X 隐性遗传病,患病多为男性,女性携带者一般不患病,但国外也曾报道过女性患病个案[14]。到目前为止,国内外报道AHDS 的患者中,SLC16A2 基因的变异已经超过100 个,包括错义、无义、缺失、插入和剪接位点突变等[15-18]。根据本例患儿病史,患儿外祖母、母亲、姐姐无临床症状,因地域原因未对患儿外祖母、姐姐进行基因家族系验证,不能获取更多临床信息进行验证,但符合X 染色体隐性遗传规律,男性半合子患病,符合相关报道[2]。
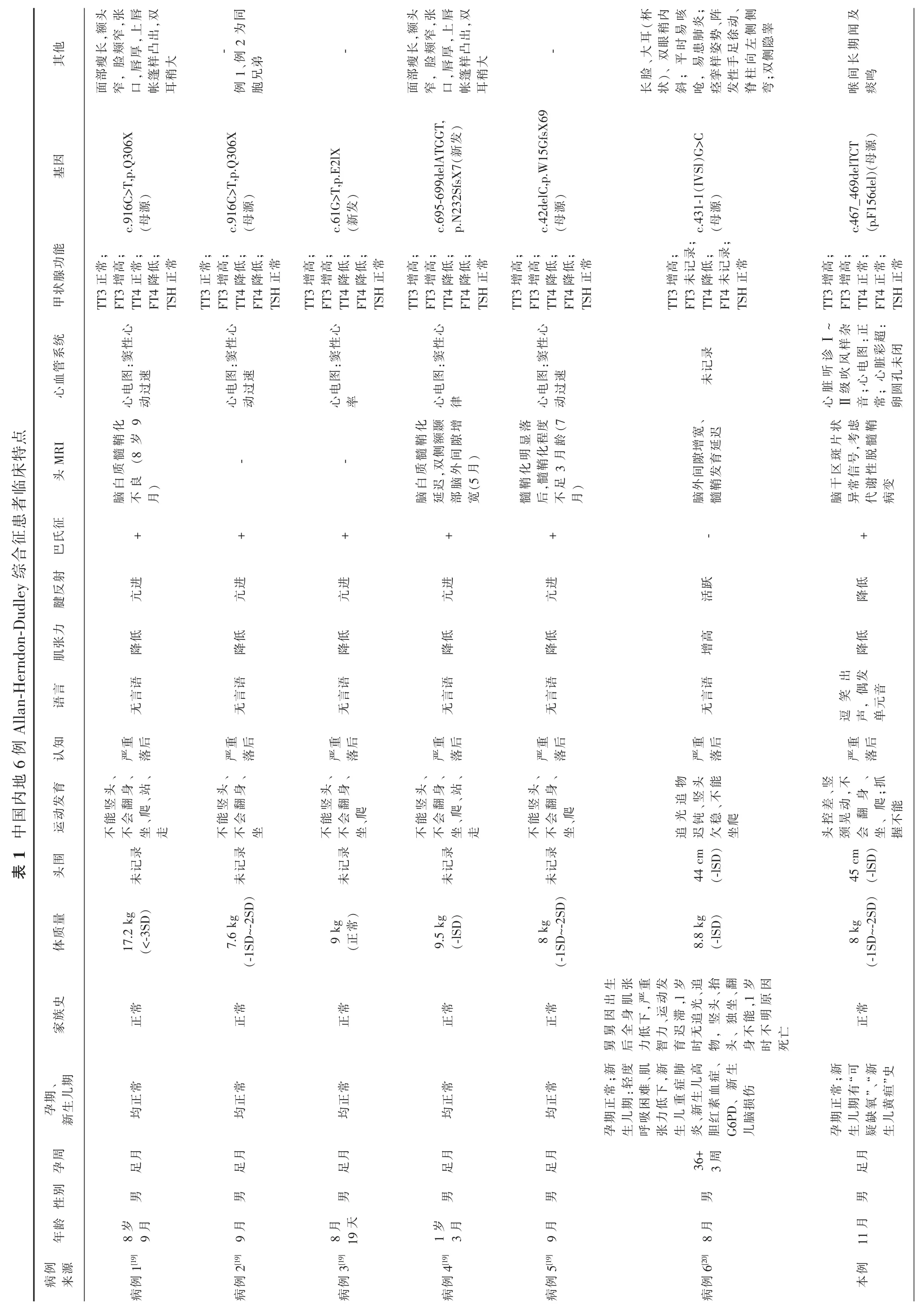
以 “SLC16A2 基因”、“Allan-Herndon-Dudley syndrome”、“AHDS”、“Monocarboxylate transporter 8”、“MCTT8”等为关键词查阅PubMed、SCIE、Springer 及中国知网、万方、维普等数据库自建库至2020 年6 月相关文献,共检索出国内已报道文献2 篇。参考文献[19-20]报道6 例内地SLC16A2 基因变异致AHDS 确诊患者,年龄范围8 月龄~8.75 岁,来自5 个家庭,均为男性,其中2 例患儿为同胞兄弟。患者父母均否认近亲结婚,5 例患儿均无明确家族遗传病史,1 例患儿疑似家族遗传史,1 例有早产、新生儿期轻度呼吸困难、肌张力低下、新生儿重症肺炎、新生儿高胆红素血症、葡萄糖-6 磷酸脱氢酶缺乏症、新生儿脑损伤等病史。6 例患儿巴氏征阳性,5 例患儿头颅MRI 提示髓鞘化异常,3 例患儿有窦性心动过速,4 例患儿有特殊面容。以上6 例患儿与本例患儿均存在严重认知、运动、语言发育落后,均存在肌张力、腱反射及甲状腺功能异常等。以上6 例AHDS 患儿与本例患儿的临床表现和SLC16A2 基因变异类型具体见表1。
多数AHDS 患者预后差,呼吸、胃肠和骨科疾病是该病最常见的并发症,其中以肺部感染最为常见,可能导致过早死亡,少数病例可存活至成年[3]。治疗上,国外研究者使用左旋甲状腺素或联合丙硫氧嘧啶、左旋甲状腺素替代激素疗法等治疗但并无明显疗效[21-26]。目前,国外研究热点为寻找可以绕过MCT8 进入大脑及特定神经细胞内的甲状腺激素[27-31]。有学者认为Triac 可能具有促进患者神经发育、改善神经功能,同时减轻外周高甲状腺素毒性的作用[32-36]。该病目前尚无特效药物治疗,可予物理、言语治疗等康复治疗。Remerand 等[3]对24 例AHDS 患者进行研究,结果显示每例患者均接受平均每周3.1 次康复治疗,其中接受物理治疗22 例、言语治疗10 例、心理治疗14 例、职业治疗6 例、矫正治疗7 例、精神病治疗3 例,之后11 例患者在人工辅助下正常上学,12 例法国患者被录取到医学教育机构。因此,虽然目前对该病尚无特效治疗药物,早期综合康复治疗非常重要。
综上所述,临床医生应加强对AHDS 的认识,根据早期可疑临床表现提高警惕并进一步完善相关检查,条件允许可进行基因检测,避免误诊漏诊。该病主要发病在男性,女性携带者一般无临床症状,但国外有女性患病报道,仍需警惕。若有明确家族史或疑似女性杂合子,应完善基因检查并接受产前遗传学咨询及胎儿期检查。但目前该病尚无特效治疗,药物治疗可能仍将是未来研究的重点,而且康复治疗也非常重要。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
中华家教(2018年7期)2018-08-01 06:32:38
特别健康(2018年2期)2018-06-29 06:13:44
文学少年(原创儿童文学)(2016年16期)2016-02-28 17:50:19
百科知识(2015年18期)2015-09-10 07:22:44
中国当代医药(2015年7期)2015-03-01 02:01:09
中国卫生(2014年6期)2014-11-10 02:30:50
教育生物学杂志(2014年1期)2014-03-11 17:06:43
中国中医药现代远程教育(2014年22期)2014-03-01 04:33:14
