
Review of the Current Synthesis and Properties of Energetic Pentazolate and Derivatives Thereof
2021-01-25 07:53:58DominiqueWozniakDavinPiercey
Engineering 2020年9期
Dominique R. Wozniak, Davin G. Piercey*
Department of Materials Engineering & Department of Mechanical Engineering & Purdue Energetics Research Center, Purdue University, West Lafayette, IN 47907, USA
Keywords:Explosives Pentazoles High-nitrogen compounds
A B S T R A C T The latest member of the azole family, the pentazolate or cyclo-N5-, has received increased attention since its first mass-spectral detection by Christe et al.in 2002.As it is carbon-and hydrogen-free,the pentazolate anion can release large amounts of energy while simultaneously decomposing to environmentally friendly nitrogen gas. Due to these attractive qualities, cyclo-N5- and related compounds are essential in the advancement of high-energy-density materials (HEDMs) research. This review aims to provide a consolidated report on all research done on cyclo-N5-,with a focus on pentazoles as energetic materials and on their experimental synthesis.Included in this review are the following:①the historical significance of cyclo-N5-; ②precursors of cyclo-N5-; ③synthesis routes of cyclo-N5- with a focus on arylpentazole precursors;④factors affecting the stability of cyclo-N5-;⑤energetic performances of current energetic cyclo-N5--containing compounds; and ⑥future possible experimental research. This review is a comprehensive summary of the current understanding of cyclo-N5-, in an effort to further understand the potential of this anion for adoption as a powerful and environmentally friendly nextgeneration explosive.
1. Introduction
High-energy-density materials (HEDMs) have attracted the attention of researchers for their extensive use in explosives, propellants, and pyrotechnics. The ever-increasing need for safer,more efficient, and more environmentally friendly HEDMs has been influential in the search for these unique materials. As such,research on the synthesis and characterization of this class of materials has come a long way,both experimentally and computationally [1,2].
‘‘Classical”explosives such as 2,4,6-trinitrotoluene(TNT),1,3,5-trinitro-1,3,5-triazacyclohexane (RDX), and 1,3,5,7-tetranitro-1,3,5,7-tetraazacycloocatane (HMX) derive their explosive properties from the combination of fuel and oxidizer in the same molecule.‘‘Second-generation” explosives such as 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurzitane (CL-20) and octanitrocubane(ONC) possess greater energy content than classical explosives due to the cage strain present in the molecule [1-3].
One of the latest thrusts in HEDM design is high-nitrogen and polynitrogen compounds. All high-nitrogen species derive their energy content from the high heat of formation of extended single- and double-bonded nitrogen systems. Nitrogen chemistry differs from carbon chemistry; unlike with carbon, where increasing bond order leads to lower average bond energy, increasing bond order with nitrogen leads to higher average bond energy[4]. Because of this novel property, the formation of nitrogennitrogen triple bonds in dinitrogen gas from single and double bonds releases a large amount of energy.This large energy release is attractive for application in HEDMs,but also means that polynitrogen compounds are metastable, making their synthesis and characterization difficult; before 2016, only N5+and the azide anion had been synthesized on a macro scale [5,6].
Recently, interest in cyclo-N5-, the pentazolate anion, has increased significantly.Before 2016,several computational studies accurately predicted the stability and existence of the pentazolate anion. Cyclo-N5-was theorized to decompose into N3-and N2in an exothermic reaction,releasing 14.3 kcal·mol-1(1 kcal=4.184×103J) of energy. The barrier of this reaction was calculated to be 27.2 kcal·mol-1[7,8].The heat of formation(ΔHf, 298 K)and ionization potential were calculated to be 59.6 kcal·mol-1and 5.06 eV,respectively [8-10]. This data is indicative of the possibility of isolating cyclo-N5-under the right conditions, and was the initial motivation of many synthetic chemists in their effort to obtain and understand this anion [11].
The mass-spectral detection of cyclo-N5-in tetrahydrofuran(THF)by Bazanov et al.[12]in 2016 was the first of several significant advances in the synthesis of cyclo-N5--containing compounds.Within three years, the synthesis of bulk cyclo-N5-, its metallic salts, and its organic salts all came to fruition [13-22]. Wang et al. [2] previously provided an extensive review of both theoretical and experimental results; however, as this field has experienced a rapid increase in interest, it is of the utmost importance to provide researchers with the most accurate and up-to-date information possible. With this in mind, we wish to add to this review with new studies and clarifications,with a focus on the synthesis and properties of the known pentazolates.Herein,the rationale behind and the synthesis of cyclo-N5-and its related compounds are described.
2. Precursors to cyclo-N5-
The experimental pursuit of polynitrogen species, specifically cyclo-N5-, has been an extensive effort spanning two decades[23]. Even with this extensive effort, only two validated experimental pathways exist for the synthesis of cyclo-N5-(Fig. 1) [2].The current most popular pathway is the reaction of a substituted aryldiazonium salt with an azide anion to yield a substituted pentazene intermediate, which then cyclizes to form a substituted pentazole precursor that can be used for the preparation of pentazolates [24-26]. The second, less favored, pathway involves the direct production of cyclo-N5-from N3-and N2, but this process involves extreme conditions of high pressure and temperature reactions in diamond anvil cells, and is less feasible for bulk synthesis than the first route. Unfortunately, the production of arylpentazole (ArN5) precursors is still a difficult task. The degree of the aromaticity of the cyclo-N5-ring,which is vital for its stability, is dependent on the substituents attached to the aryl group.Furthermore,cleavage of the pentazolate anion from the ArN5precursor without destroying the pentazole ring has proven to be difficult. Consequentially, extensive theoretical work has been performed to determine the optimal conditions and precursors for the synthesis of unsubstituted pentazole [2,24-29].
2.1. Aryl- and benzyl-substituted pentazoles
The simplest substituted ArN5is simple phenylpentazole(PhN5). The first experimental attempts toward this class of compounds were undertaken by Noelting et al. [30] in 1892, but only aryl azides were produced. The first successful synthesis of ArN5is often credited to Huisgen and Ugi, but should actually be attributed to Clusius and Hürzeler[31]in 1954.Shortly afterward,Huisgen and Ugi[32,33]were able to confirm the existence of ArN5by gas-volumetric and kinetic studies.Physical confirmation of the structure of ArN5was achieved in 1983 by Wallis and Dunitz[34], who reported the first X-ray-measured crystal of pdimethylaminophenylpentazole (DMAP-N5)—a pentazole whose decomposition at room temperature was further explored by Yang et al. [35].
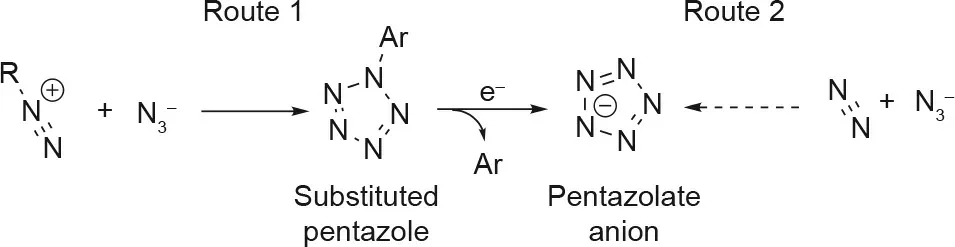
Fig.1. Synthetic routes to cyclo-N5-.An ArN5 intermediate is shown in Route 1, as it is the most common substituted pentazole, but other substituted pentazole intermediates have also been attempted.
The instability of PhN5has largely prohibited its further experimental use as a precursor to other pentazoles. Computational and experimental studies have revealed that ArN5is thermally stabilized by electron-donating groups in the para and ortho positions to the pentazole ring on the benzene ring and destabilized by electron-withdrawing groups at those same positions[28,29].Aryl substituents meta to the pentazole ring also destabilize the ArN5.These results are explained through the resonance between the phenyl groups and the pentazole ring, with stability increasing with increasing resonance [28,29]. The bond dissociation energy of the C-N bond in substituted ArN5was found to be 362-402 kJ·mol-1, but the bond dissociation energy of pentazole (N-N bonds) was calculated to be 109-117 kJ·mol-1, meaning that the pentazole ring would fall apart before it could be cleaved from its substituent(Fig.2)[2,29].These same effects on the intermediate arylpentazene were explored in 2009 by Burke and Fazen [25]and again in 2019 by Ren et al.[24].These studies emphasized the importance of electron-donating substituents for the formation of the trans-cis isomer, the only pentazene intermediate that is able to successfully cyclize into the arylpentazene. Ren et al. [24] also provided detailed reaction pathways of the possible pentazene intermediates and described the influence of substituents on the cyclization of the pentazole over the loss of N2.
PhN5substituted with singular alkali metals lithium (Li),sodium(Na),and potassium(K)at the ortho,meta,or para positions was also computationally explored [36]. It was found that the metal-substituted PhN5derivatives had a significantly improved energy barrier toward decomposition of the pentazole ring when compared with the -OH, -OCH3, and -N(CH3)2substituted PhN5compounds of Zhang et al. [29]. Also observed was a metal-nitrogen bond between an ortho-metal substituent and the nitrogen ring. This interaction was not present for the para- and metametal-substituted compounds(Fig.3)[36].The strong intramolecular interactions of the ortho-substituted compound effectively impose a more negative charge on the pentazole ring. This increased negativity means that the dispersion interactions are also increased, leading to greater stability of the pentazole ring[36].
Benzylpentazole (BnN5)and its derivatives have also been considered as precursors to cyclo-N5-[2,37].Zhang et al. [37]computationally studied the effects of electron-withdrawing and electron-donating substituents on the stability of the pentazole ring and the C-N bond. Electron-withdrawing groups simultaneously lowered the stability of the pentazole ring and the C-N bond.In comparison with ArN5precursors,BnN5and its derivatives possess more stable N5rings and less stable C-N bonds[28,37].At first glance, it may seem that BnN5would be the better choice of precursor to the unsubstituted pentazolate anion; however, due to the extreme instability of the necessary benzyldiazonium synthetic intermediate, BnN5would be difficult or near impossible to produce experimentally [38].
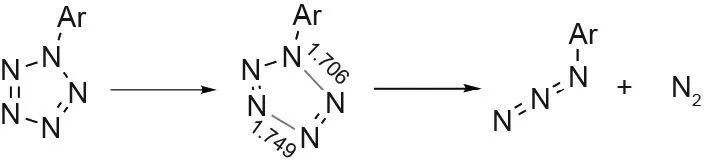
Fig. 2. Decomposition of ArN5 (unit: Å).
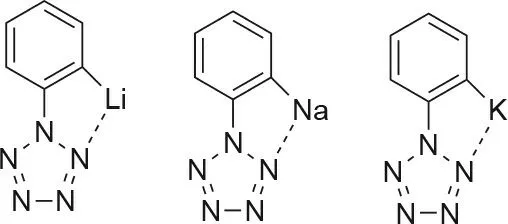
Fig. 3. Metal-pentazole interaction of ortho-substituted alkali metals Li, Na, and K phenylpentazoles [36].
2.2. Heterocyclic-substituted pentazoles
Tetrazolylpentazole was the second substituted pentazole to be produced [39]. As it decomposes at -50 °C, nuclear magnetic resonance (NMR) studies performed at -70 °C were the only confirmation of its existence. Computational methods revealed that the tetrazole had similar effects on pentazole conjugation as an aryl group. When the anionic tetrazolepentazole substituent was compared with its neutral counterpart, the activation energy for decomposition increased [39].
Six-membered heterocycle pyridylpentazole (PyN5) and its derivatives were also considered as potential precursors to pentazole by Zhang et al.[40].Replacing the aryl group of PhN5with pyridyl lowers the activation energy of pentazole N-N dissociation(8.4-30.8 kJ·mol-1)and that of the C-N bond(11.3-21.0 kJ·mol-1)[40]. The decrease in stability of the pentazole ring is more significant than that of the C-N bond; therefore, PyN5is not a better precursor for cyclo-N5-than the aryl pentazoles [40].
2.3. Other substituted pentazoles
The pentazole ring’s stability is dependent on its aromaticity.Since a nitrogen-nitrogen triple bond is the most stable conformation, compared with a double or single bond, increasing the aromaticity helps to force a more double-bonded nature between all of the nitrogen atoms in the ring, thus increasing the stability[41]. The ability to separate the σ- and π-electron systems also lends to the stability of the pentazole ring[42].Due to these properties, the pentazole ring is a relatively stable polynitrogen compound, and can potentially be used as a building block for other polynitrogen systems. Polynitrogen (Nn) systems from n = 5 to n = 60 can be split into several different groups, depending on the number of nitrogen atoms and their relative stabilities versus conformation(Figs.4 and 5(a))[8,41,43-46].In general,structures containing multiple pentazole rings are more stable than the corresponding open-chain structures,as found in N18,N20,and N30,with the N8azidopentazole and N10bispentazole structures representing energy minima(Figs.4 and 5(a))[47,48].It must be noted that the highly sought after N5+N5-compound was computationally determined to be unable to exist (Fig. 5(a)) [8,47].
Other substituted pentazoles including methyl,cyano,amino,or N-oxide groups have been computationally predicted to be stable compounds with high performances (Figs. 5(b)-(d)) [43-46,49]. A more recent computational study on pentazole derivatives with oxygen balances of zero or higher was also conducted (Fig. 6)[49,50].Many of these compounds are predicted to be good candidates for HEDMs, although the necessary synthetic methods are not yet available to allow their experimental synthesis.
3. Methods of synthesis of cyclo-N5-
So far, there are only two methods of producing cyclo-N5-(Fig. 1). The most experimentally convenient route is to cleave the pentazolate ion from substituted aryl pentazole precursors[2]. In order for these precursors to be effective, three considerations must be made: ①Electron-donating substituents must be placed at the para position of the benzene ring relative to the pentazole ring in order to force as much electron density into the pentazole ring as possible, which increases the stability of the pentazole ring and weakens the connective C-N bond; ②the CN bond must be cleaved while leaving the N-N bonds of the pentazole ring intact; and ③an effective analytical method must be used to detect the pentazolate anion.
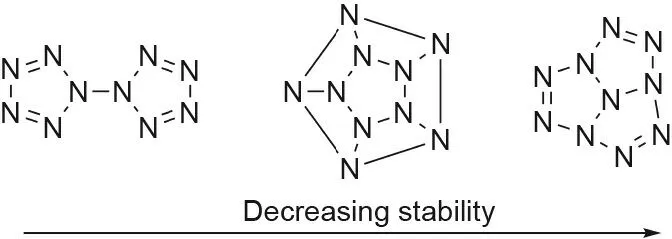
Fig. 4. Polynitrogen compounds containing ten nitrogen atoms in different conformations. The stability of the compounds decreases from left to right.(Compounds from Ref. [41].)
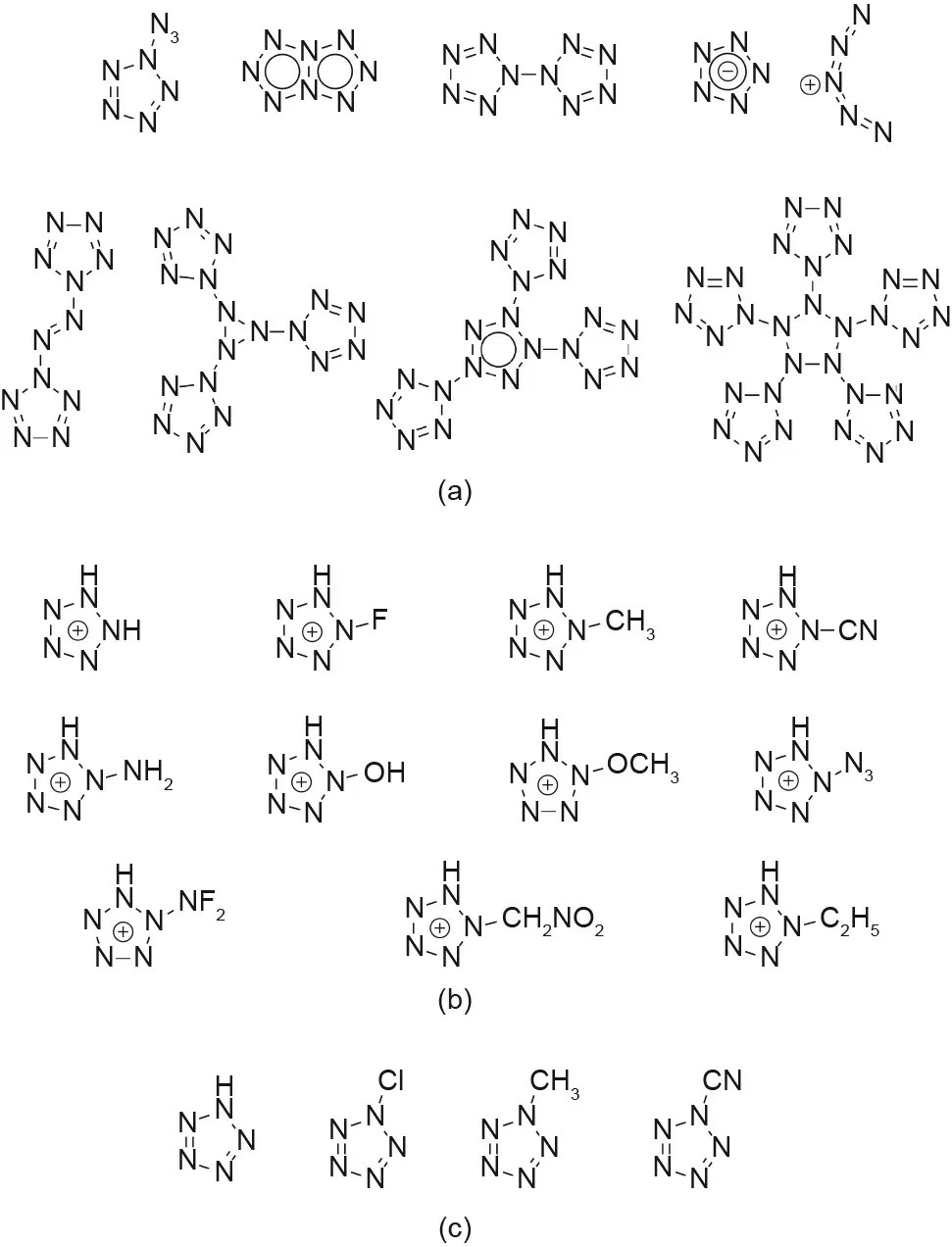
Fig. 5. Other computationally explored substituted pentazole compounds. (a) N5-based polynitrogen; (b) cationic-substituted pentazoles (R-N5H+); (c) neutral substituted pentazoles (R-N5). (Compounds from Refs. [8,43-46].)
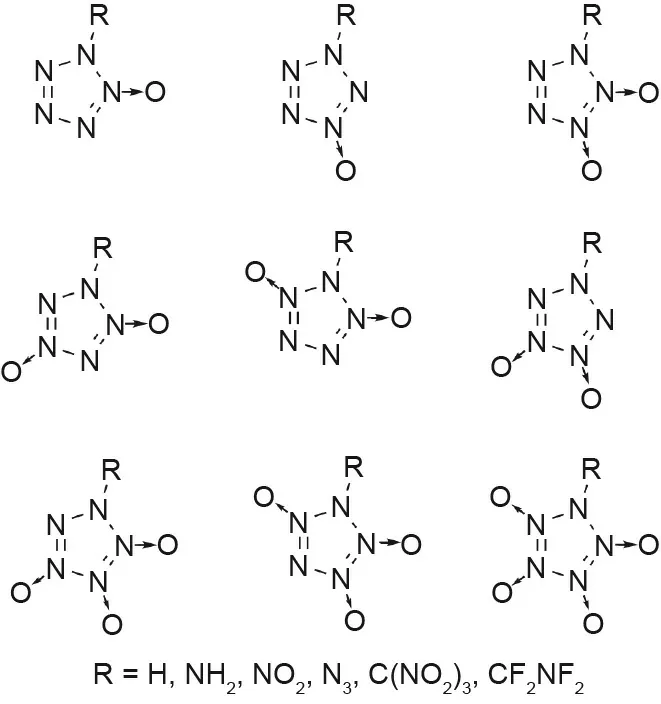
Fig. 6. N-oxide-containing pentazole derivatives. (Compounds from Refs. [49,50].)
3.1. Cleaving the PhN5 C-N bond through electronic excitation
Mass-spectral studies by Christe and Östmark, along with a computational study by Haas, have revealed a great deal about the mechanism of C-N cleavage in ArN5systems. Christe’s study used tandem mass spectrometry (MS/MS) with collision-induced dissociation (CID) at both high and low collision voltages on phydroxy and p-dimethylamino phenylpentazoles. It was discovered when studying p-hydroxyphenylpentazole that, at low voltages, the parent anion decomposed by the loss of two N2groups and, finally, the loss of CO. At high voltages, the C-N bond was instead cleaved to yield the pentazolate anion fragment, which then decomposed into the azide anion and neutral N2(Fig.7)[23].
Östmark et al. [51]performed the same experiment using laser desorption ionization (LDI) time-of-flight (TOF) MS on DMAP-N5,and employed density-functional theory(DFT)analysis to decipher the fragmentation pathway. Calculations showed that production of the DMAP-N5radical through electron attachment followed by decomposition into the pentazolate anion and the DMAP radical was the preferred pathway (Fig. 8) [51]. It was also found that the activation energy for C-N bond cleavage was higher than that for decomposition of the pentazolate ring by 1.6-6.6 kcal·mol-1[51]. At higher laser power, it was observed experimentally that,although more fragmentation was seen,C-N bond cleavage always occurred before decomposition of the pentazolate anion. Östmark et al. [51] further concluded that DMAP-N5could be excited in a way that encouraged C-N bond cleavage, but the production of significant amounts of cyclo-N5-was not possible by this method.
The contrasting results obtained by Christe and Östmark were explained by a computational study by Belau et al.[52].The study compared the p-hydroxyphenylpentazolate anion, a closed-shell system, with an open-shell DMAP-N5anion radical equivalent,and reported that the unexpected result for the high energy cleavage of hydroxyphenylpentazole can be explained by the electronic states of the reactants and products. The hydroxyphenylpentazole anion experiences negative charge transfer from the oxygen to the pentazole ring when the C-N bond is stretched,while the negative charge in the DMAP-N5anion radical remains on the pentazole ring during the excitation process[52].It was also observed that conical intersections exist for both species, allowing the formation of the pentazolate anion from the excited state (Figs. 9 and 10). The hydroxyphenylpentazole system was able to produce the pentazolate anion only at high voltages because the excited state that was created was able to pass through a conical intersection to the corresponding ground state; however, this was not the case for the DMAP-N5system.The difference lay in the barriers of C-N cleavage and N2extrusion.The DMAP-N5anion radical had similar barriers,but the hydroxyphenylpentazole anion had an increased barrier to C-N bond cleavage and a decreased barrier for N2extrusion [52].The same result was obtained by Portius et al. [53] when using laser excitation along with infrared spectroelectrochemistry to study the excited state of DMAP-N5.
Several attempts were made at producing the pentazolate anion with photochemical methods by Haas’group[54-57],using Raman and ultraviolet-visible (UV-Vis) spectroscopy as the analytical methods. Various experiments were performed under a variety of conditions and excitation modes. It was found in all cases that if any cyclo-N5-was formed,it was only in trace amounts.The major products were decomposition products of the precursor [54-57].
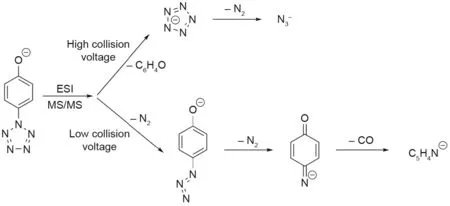
Fig. 7. Fragmentation of p-methoxyphenylpentazole as observed by Vij et al. [23]. ESI: electrospray ionization.
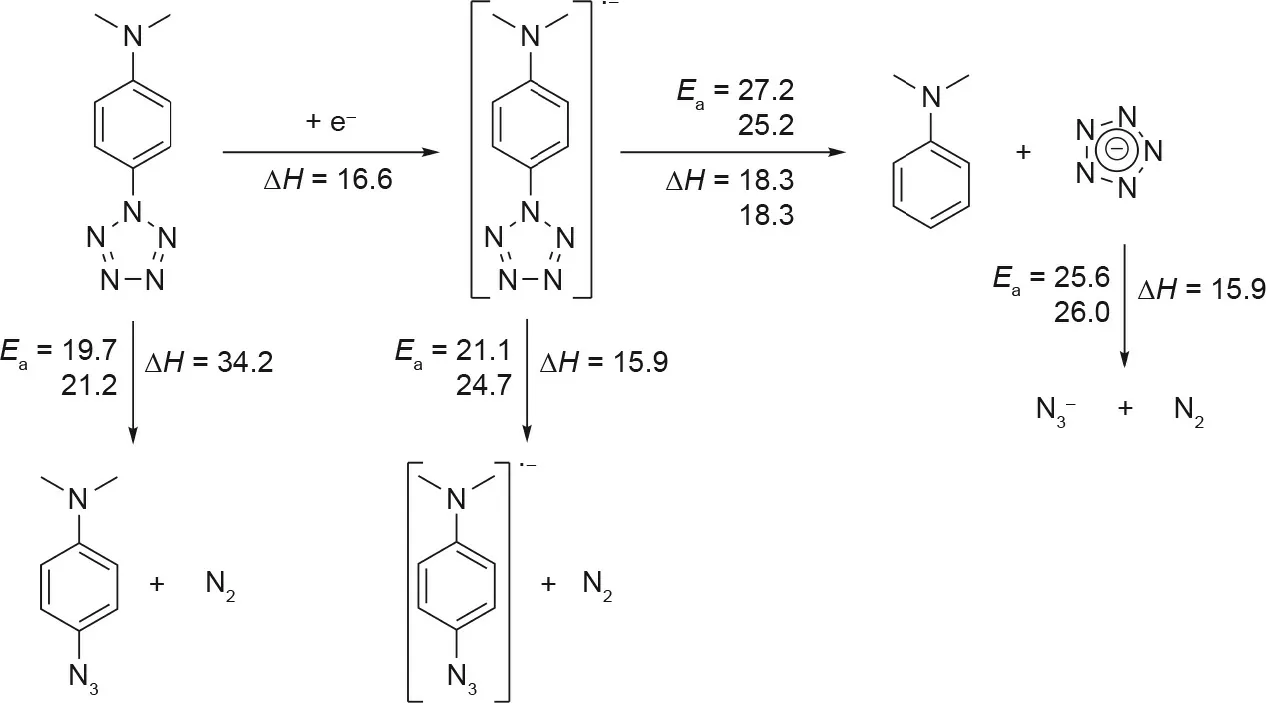
Fig. 8. Proposed decomposition pathway for DMAP-N5, as studied by Östmark et al. [51]. Activation energies (Ea) and enthalpies (ΔH) are at 298 K and in kcal·mol-1.
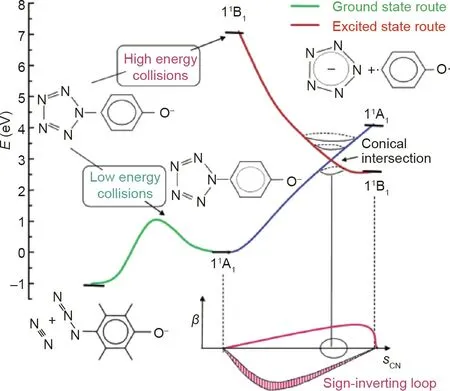
Fig.9. Energy levels of p-hydroxyphenylpentazolate anion created by high and low collision voltages. sCN stands for the C-N stretch coordinate; β stands for upward bend (+β) or downward bend (-β). Reproduced from Ref. [52] with permission of the American Chemical Society, ©2004.
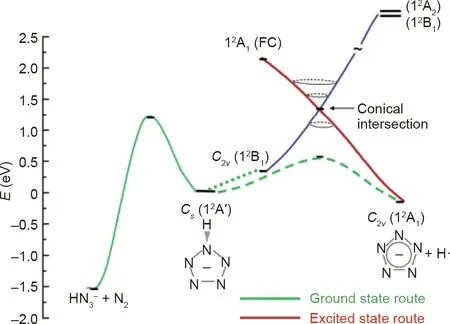
Fig. 10. Dissociation scheme of the DMAP-N5 radical anion equivalent system. FC stands for Franck-Condon excitation.Reproduced from Ref.[52]with permission of the American Chemical Society, ©2004.
3.2. Redox methods for cleavage of the C-N bond
Butler et al. [58] attempted the redox cleavage of ArN5and the capture of the pentazolate anion using ceric ammonium nitrate(CAN), a single-electron transfer (SET) reagent, and zinc nitrate.They reported that the unique15N NMR signal at (-10.0 ± 2.0)ppm obtained from the reaction was ZnN5(NO3). This was the first published claim of an isolated pentazolate. This claim was later refuted by Schroer et al. [59], which prompted Butler et al. to repeat the experiment with various ArN5compounds substituted with one, two, or three15N atoms [60]. Under these conditions,only decomposition products of N5-were recovered [60].
Bazanov et al. [12] are credited with the first successful direct detection of the pentazolate anion in THF solutions using alkali metals to reduce ArN5at low temperatures. The alkali metal was able to produce the PhN5radical anion from the parent PhN5,which was allowed to warm to room temperature to produce the cyclo-N5-[12].The pentazolate anion was detected by electrospray ionization (ESI) MS with a signal at m/z = 70. Isotopic labeling of one or two nitrogen atoms produced the corresponding m/z = 71 and 72. MS/MS analysis of the signal at m/z = 70 showed a peak at m/z=42,corresponding to the azide anion,an expected decomposition product of pentazolate. The pentazolate anion was indefinitely stable when stored at below -40 °C, but was only stable for a few minutes at room temperature [12].
Bazanov et al.[61]later repeated this experiment in an attempt to optimize the reaction conditions. When using pure sodium metal in pure solvents no pentazolate anion was formed.However,the use of oxide-passivated (solid electrolyte interface) sodium produced the highest amounts of pentazolate anion[61].This reaction methodology is unfortunately limited by its long reaction time; the shortest reaction time was two weeks and the longest was three months. Because of the slow reactions times and yields that were only sufficient for detection and not for bulk isolation,Bazanov et al.[61]concluded that sodium was too strong a reducing agent.
The most successful example of the production of the pentazolate anion through redox chemistry was by Zhang et al. [13], who used a ferrous bisglycinate and m-chloroperbenzoic-acid system to cleave the pentazolate anion from PhN5in 19%yields,in the first and largest-yielding bulk synthesis of a pentazolate species.The pentazolate product produced was initially said to be(N5)6(H3O)3(NH4)4Cl, in which the pentazolate anion remained unprotonated; this was strongly debated by several groups[62-65]. Huang and Xu [62] reasoned that the O2ellipsoid in the asymmetry unit plot appeared to indicate that the electron density of the proposed compound was less than should be assigned. The inconsistency in the symmetry of the proposed compound and the symmetry of the studied infrared spectrum(C2vcompared with D5h)was also questioned.Using DFT calculations,it was suggested that the pentazolate anion was actually protonated by the H3O+and was more likely to exist as HN5[62].Jiang et al.[63]responded to this suggestion by insisting that their high-quality X-ray crystallographic data, along with NMR and DFT thermodynamic studies,were more accurate and stronger evidence for the proposed unprotonated pentazolate product. It was later noted by Chen et al. [64]that Zhang et al. had not considered how external conditions such as temperature and solvent could greatly impact proton transfer in the solid versus liquid phases. Chen et al. [64] used ab initio molecular dynamics (AIMD) simulations to more accurately predict the behavior of the cyclo-N5-in the(N5)6(H3O)3(NH4)4Cl crystal and dimethyl sulfoxide (DMSO) solvent. It was found that, in the solid, the cyclo-N5-always coexisted with some amount of its protonated counterpart, with a maximum of 23% HN5being present at 228 K. Conversely, it was found that, in DMSO, the solvent and temperature effects narrowed the proton affinity difference between cyclo-N5-and water or ammonia [64]. Further DFT and AIMD studies by Huang et al.[65]also supported the preferred existence of HN5. Lu et al. eventually settled the discussion in a recent paper†† This paper has not been published., by suggesting that the original crystal structure was disordered, which had resulted in its misinterpretation. They concluded that the compound produced was in fact (NH4)7(N5)6Cl and not(N5)6(H3O)3(NH4)4Cl.Regardless of the debate,the 19%yield obtained in the original study by Zhang et al. [13] is still the best yield of pentazolate salt to date. The salt produced was also stable at room temperature and decomposed at 116.8 °C.
This experimental success is, without question, what has allowed the synthesis of many other pentazolate-containing species in the few years since its publication. Unfortunately, the mechanism for the C-N bond cleavage is not yet entirely thoroughly understood, which impedes the development of more efficient methods of synthesis.In 2018,Yu et al.[66]computationally explored the pentazolate formation mechanism of this experimental system in hopes of answering this question.Although they were able to conclude that proton transfer from the radical anion was not kinetically favorable for formation of the pentazolate, they were unable to clearly explain the C-N bond cleavage. In 2020,Shang et al. [67] proposed a reaction mechanism for the cleavage of the pentazolate anion. They concluded that metachloroperoxybenzoic acid (m-CPBA) was not responsible for the activation of the C-N bond cleavage, but was instead responsible for the oxidation of [Fe(Gly)2]. This oxidation in turn generates a high-valent iron(IV)-oxo product, which is the key component for C-N cleavage and generation of the pentazolate anion. They noted in their study that the formation of this complex is heavily dependent on low reaction temperatures and that the solution will produce a reddish-brown precipitate when the temperatures are too high [67]. They concluded that higher yields of pentazolate may be achieved by experimentation with the ligands of the iron(IV)-oxo complex [67].
3.3. N3- + N2 reaction at high temperature and pressure
The other currently known synthetic route to cyclo-N5-was carried out by Oleynik’s group[68-71],who produced the pentazolate anion by heating liquid nitrogen and azide salts in diamond anvil cells and laser-heating to high temperatures and pressures.This method allows sufficient energy to overcome the barrier to N3-+N2cyclization, while the high pressure serves to stabilize cyclo-N5-. Unfortunately, upon release of the pressure, no cyclo-N5-could be detected [68-71]. Laniel et al. [72] performed a similar experiment and was able to detect cyclo-N5-at ambient pressure,but only in an inert atmosphere.Laniel et al.[73]also explored the Li-N phase diagram resultant from pure lithium in excess nitrogen compressed and heated to a maximum pressure and temperature of 73.6 GPa and 2500 K, respectively. Through this method, they were able to obtain Li3N, LiN, LiN2, and LiN5and examine their respective pressure stability domains [73]. Theoretical highpressure synthesis of metal pentazolate-containing compounds was also recently conducted, predicting copper, magnesium, and barium compounds that are metastable under ambient conditions[74,75].At the present time,cleavage of ArN5precursors is still the best route to the cyclo-N5-for laboratory-scale preparation.
4. Stability and bonding of cyclo-N5-
Experimentally, the pentazole anion was determined to have C2vsymmetry with differing N-N bond lengths. This differs from the D5hsymmetry with equivalent N-N bond lengths that was predicted through computational methods[13]. It was found that the lone pairs on the nitrogen are able to form bonds with the cations surrounding them, causing the N-N bonds to change in length.Although different cations can bind to the nitrogen of the pentazolate ring, all of the reported bond lengths are within 1.30-1.33 Å,which is an intermediate length between those of a single and double bond. It was also found that cyclo-N5-retains its aromaticity when properly stabilized through intramolecular interactions[13,76].
4.1. Hydrogen bonding cyclo-N5- complexes
The metal-pentazolate hydrate complexes (Na, Mn, Fe, Co, Mg,and Ba) experimentally exhibit a heavy dependence on water for their stability [13,77]. A more recent computational study also found that water plays an important role in the stabilization of metal-pentazolate hydrates [78]. This function is due to water’s ability to act as both a hydrogen-bond donor and acceptor[12,78]. In all cases, water molecules stabilize the complex either through coordination(coordination water)to the metal or by forming hydrogen bonds to the pentazolate anion (constitution water)(Fig. 11) [78].
Interaction with the lone pairs on water molecules causes the energies of the metal 4s and 3d orbitals to increase and subsequently form a dative (or coordinate) bond [78]. This in turn decreases the interaction of the metal and pentazolate anion by decreasing the electron-withdrawing effect of the metal,increasing the activation energy for the extrusion of N2. It was also observed that water induced a steric hindrance on the metal-pentazolate complex that limited the pentazolate anions’direction of coordination [13].
In contrast to coordination water, constitution water forms hydrogen bonds with two nitrogen atoms of separate cyclo-N5substituents in metal-pentazolate complexes (Fig. 11). Upon the removal of a constitution water molecule from the metal-pentazolate complex, there is a marked decrease in the activation energy for N2extrusion.Thus,the loss of constitution water leads to instability in the compound and is the first step in decomposition [78].It must be noted that coordination water molecules were found to have a more significant effect on the stability of a metal-pentazolate hydrate complex than coordination waters[78].In that study,coordination water caused the kinetic barrier to decomposition to increase by an average of 7.6 kcal·mol-1, while constitution water caused the kinetic barrier to decomposition to increase by an average of 7.3 kcal·mol-1[78].
For the non-metal-containing organic salts synthesized by Xu et al.[19],amino groups formed hydrogen bonds with the pentazolate anions instead of water,yielding anhydrous salts.It was noted that all the salts produced had hydrogen bonds to every pentazolate nitrogen. It was also found that these hydrogen bonds were stronger than the total van der Waals interactions found in the molecule, making them significant contributors to the stability of the compound [19]. Xu et al. [79] also produced the metalfree hydrated pentazolate salts (C4H9N10+)(N5-)·3H2O and(C5H9N10+)2(Cl-)(N5-)·3.5H2O, both of which had N-H and O-H hydrogen bonding present. When comparing the thermal stabilities and Hirshfeld surface analyses of the two compounds,it was found N-H hydrogen bonding was more beneficial for the stability of the cyclo-N5-ring than O-H hydrogen bonding [79].
4.2. Acidity and stabilization of cyclo-N5-
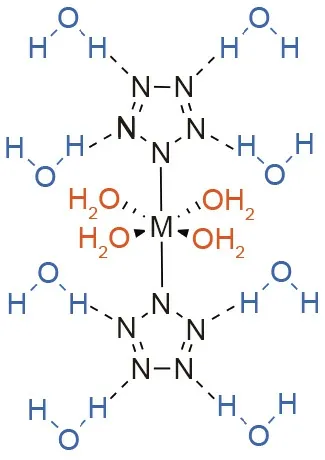
Fig. 11. Bonding modes of coordination water (orange) and constitution water(blue) in metal-pentazolate hydrate complexes.
Quantum calculations by Zhang et al. [80] revealed that the pentazolate anion has both σ and π aromaticity. The πaromaticity arises from the hybridization of the cyclic nitrogen atoms, while the lone pairs are able to spread themselves across the five-membered system, giving rise to σ-aromaticity. It was noted that this ‘‘dual aromaticity” is different from that observed in metal aromatic systems. In cyclo-N5-, these aromatic systems are separate, whereas in metal aromatic systems, they are combined [80]. These competing aromatic systems stabilize cyclo-N5-.
The study also observed that the dual aromatic system of the pentazolate anion is affected by the molar proportion of acid present in the solution. Binding to one and two hydronium ions decreases the aromaticity of the system, making it less stable.However, if three to four molar equivalents of hydronium ions can be maintained in the solution, the pentazolate anion is able to overcome the acid-base neutralization rules and form hydrogen-bonded complexes to restore its dual aromaticity [80].In this manner, an acidic environment can impart stabilization to the cyclo-N5-. This property is most likely the reason why the ferrous bisglycinate and m-CPBA cleavage and stabilization system developed by Zhang et al. [13] was so successful, as none of the other synthesis methods previous to theirs consisted of acidic reaction solutions.
4.3. Bonding in metal-cyclo-N5- compounds
The ability of various metal cations to form complexes with cyclo-N5-has been studied computationally for several years[11,81-91]. Metal-pentazolate complexes (pentazolides) have been calculated to have three possible coordination modes: monodentate (η1-N5), bidentate (η2-N5), and metallocene-like (η5-N5). Mono and bidentate coordination involve the σ-bonds of the pentazolate anion, while metallocene-like coordination involves the π-bonds (Fig. 12) [11]. η2-N5coordination was computationally observed in the pentazolides of alkali metals (Na and K) and alkaline-earth metals (Mg, Ca, Sr, and Ba), with most complexes being planar [84,85]. Lithium was the only outlier, in that it preferred η5-N5coordination [11]. Several pentazolides of transition metals and rare earth metals were also computationally studied;for mono- and bis-pentazolate-containing compounds, both η2-N5and η5-N5coordination was observed, while η1-N5was found to be favored for multi-pentazolate structures[86-89].Experimentally, the bonding modes of metal-pentazolate complexes differ from those calculated. Upon comparison with the metal-pentazolate complexes that have been experimentally produced to date,the pentazolate anion has only been observed to coordinate in a monodentate manner, where anywhere from one to all five nitrogen atoms in the ring may form monodentate bonds to up to five separate metal cations [92]. Bidentate and metallocene-like structures have yet to be observed experimentally [13-18,81,93].
5. Energetic pentazolate compounds
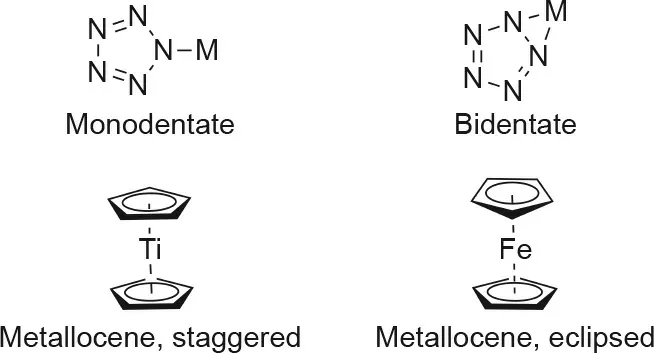
Fig. 12. Coordination modes in pentazolides.
Computational studies have predicted many stable forms of pentazolate-containing compounds, and have also attempted to predict their performance as energetic materials [8,45-50,94].These types of studies are invaluable, as they often help to guide experimentalists in their synthesis design and, in some cases, act as further confirmation of experimental findings—such as the recent work done by Christe et al.[94].While computational studies far outnumber experimental studies, several types of energetic pentazolate complexes have been experimentally obtained in the past few years.Many of these compounds were examined as energetic materials,in that the density,heat of formation,and temperature of decomposition were all determined. Using these properties, several pentazolates have had their explosive performances(i.e.,detonation pressure and velocity)predicted computationally. Pentazolate-based coordination polymers (CPs) have also been experimentally obtained [81]. In most cases,the metal-inorganic frameworks (MIFs) were more useful for the study of the bonding mode of pentazolate than they were as energetics, due to their low densities. Many of these materials were not further characterized for their detonation velocities or their sensitives.Since we are most interested in the detonation parameters of these compounds, we will only discuss the compounds that were fully characterized as energetic materials, including their detonation velocities,pressures,and sensitivities(Fig.13).The energetic properties of all of the discussed compounds are summarized in Table 1[2,16,19-22,81,96].
5.1. Thermal stabilities
Of the compounds reported here, compound 17, a threedimensional (3D) MIF, has the highest temperature of decomposition (129 °C). The structure shows that the average N-N bond length in the pentazole rings is 1.323 Å, which falls between that of a single and double bond [16]. Every pentazolate nitrogen in the compound was also bound to a sodium ion,with the five coordinated sodium ions being almost coplanar with the pentazolate ring [16]. The 3D structure resulting from the coordination of the sodium, pentazolate, and oxygen ions included ring strain, chelation, bridging actions, and electrostatic forces that are attributed to the high thermal stability relative to pentazolate-containing compounds [16].
Non-metallic energetic salts of pentazolate depend on countercations and hydrogen bonding for thermal stability [20]. For these types of compounds, decomposition of the pentazolate ring is almost always accompanied by formation of HN3gas before further decomposition occurs.This further decomposition is dependent on the counter-cations[20].Compound 11 possesses the highest temperature of decomposition of all energetic pentazolate metal-free salts (124.8 °C) [21]. Each nitrogen in the pentazolate ring is involved in a hydrogen bond with the counter-cation, and each counter-cation is very close to coplanar with one of the two adjacent pentazolate rings[21].The relatively high thermal stability of this compound is attributed to its extensive hydrogen bonding[21].The compound with the comparatively lowest thermal stability is compound 4, which has a temperature of decomposition of 80.8 °C [20]. This compound lacks hydrogen bonding capability and coordination bonds [20]. In general, most pentazolate salts decompose below 110 °C.
5.2. Mechanical stabilities
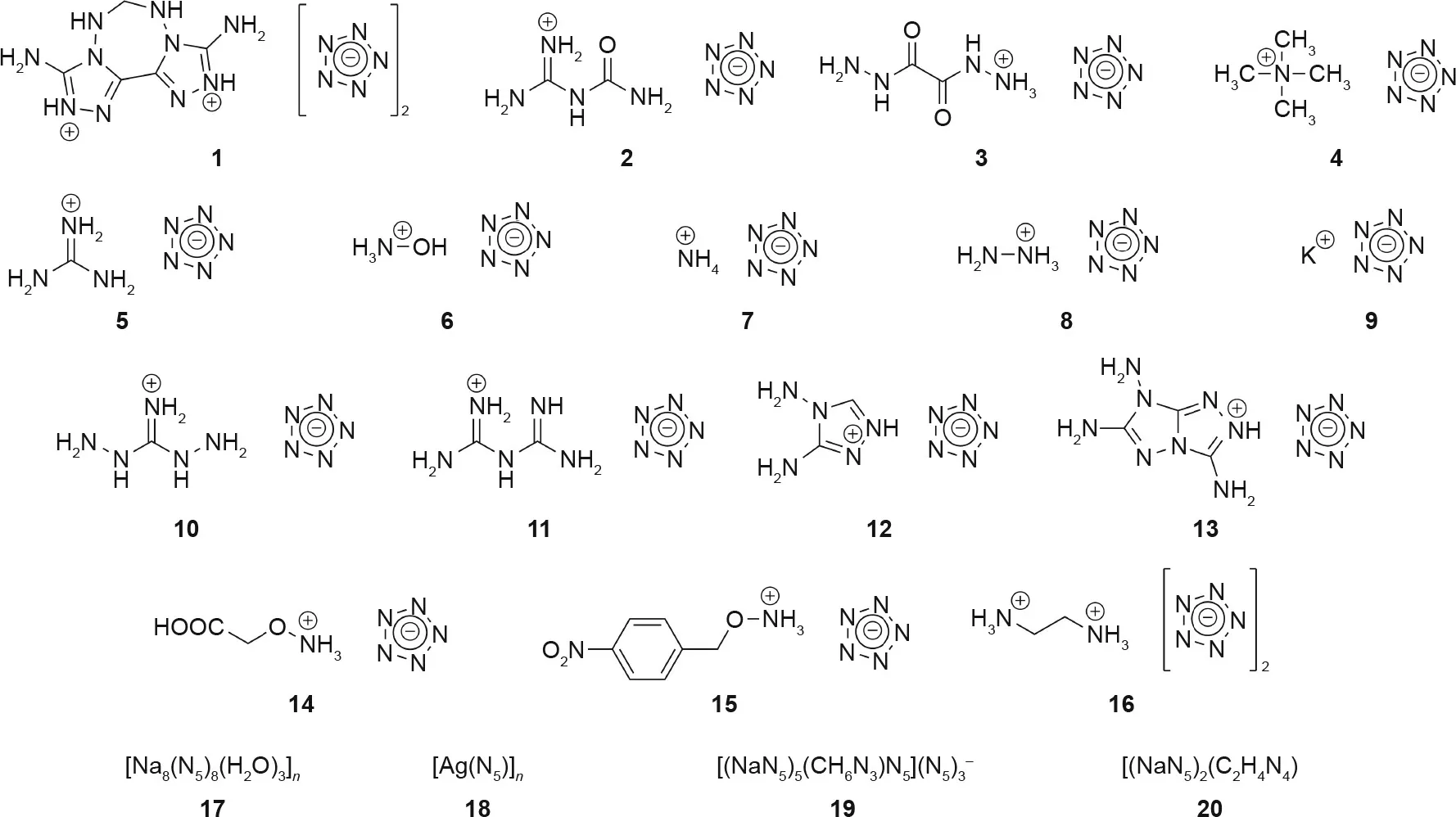
Fig. 13. Energetic pentazolate-containing compounds described in Table 1. 1-16: energetic salts; 17, 18: three-dimensional (3D) MIFs; 19, 20: 3D CPs. Structures for compounds 17-20 can be found in their respective references listed in Table 1.
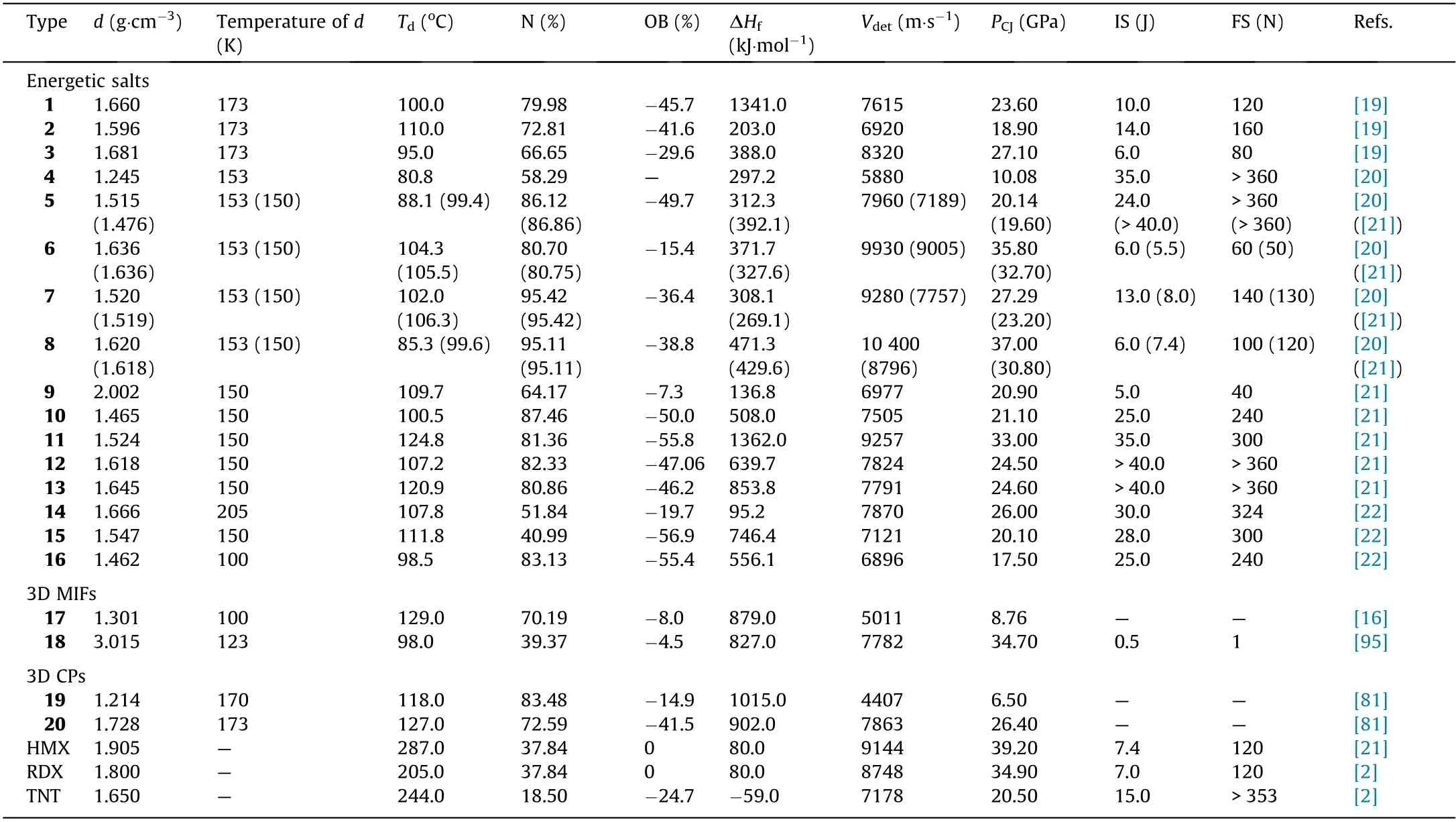
Table 1 Energetic properties of energetic pentazolate-containing compounds.
In general,mechanical stabilities—that is,the sensitivity toward friction and impact—are dependent on the intramolecular interactions within a compound [3]. Mechanical stabilities increase with increasing hydrogen bonding and layer-by-layer stacking in the solid state because these interactions are able to absorb mechanical energy and transform it to kinetic energy in the form of layer sliding [21]. Compounds 5, 12, and 13 are all classified as insensitive to impact and friction (impact sensitivity (IS) >40 J;friction sensitivity (FS) >120 N) [21]. This is due to their layerby-layer stacking patterns, in which there are π-π interactions between the layers[21].It was also noted that the hydrogen bonding and π-π interactions observed between the layers of compounds 5, 12, and 13 were much stronger than those of the other compounds studied [21]. In contrast, compounds 9 and 18 lack these abilities and,as a result,are the most sensitive to mechanical stimuli.In general,pentazolate-containing compounds are comparable to or more mechanically stable than classical explosives such as TNT, RDX, and HMX (Table 1).
5.3. Detonation performances
Detonation performance is heavily dependent on the density of the molecule and the heat of formation [20]. The 20 presented compounds all have relatively low densities; only six have densities that exceed that of TNT(1.65 g·cm-3),yet all compounds have very high heats of formation (95.2-1362.4 kJ·mol-1) compared with classical explosives (-59-80 kJ·mol-1) (Table 1), due to their increased nitrogen content. From this, we can conclude that the performances of pentazolate-containing compounds will largely reflect the heat of formation. Furthermore, in the ongoing search for new energetic pentazolates, focus should be placed on compounds with the potential for high density; a high-density pentazolate combined with the innate high heat of formation of the pentazolate anion would result in materials with very high performance. Compounds 3, 6, 8, and 11 have the highest detonation velocities, all exceeding that of HMX. Compound 11 is interesting in that the heat of formation is a result of the diguanidinium’s high heat of formation (1618 kJ·mol-1), which shows that the heat of formation of a pentazolate-containing compound can be increased by the incorporation of a cation with a high heat of formation[21].Metal-pentazolate compounds will generally have high density but low performance in comparison with nitrogen-rich pentazolate compounds. For example, compound 18 has an extremely high density of 3.015 g·cm-3but only has a detonation velocity of 7782 m·s-1,since much of its density comes from the silver cation,which lacks energy content.The same type of low performance can be seen in non-metallic pentazolate compounds with low nitrogen content such as compound 4, which has a detonation velocity of 5880 m·s-1.Of the 20 compounds presented here,13 have detonation velocities that are higher than that of TNT.
6. Conclusion
Recently, interest in the pentazolate ion and its derivatives has increased considerably as a result of the effective lab-scale preparation of a pentazolate salt from ArN5. After decades of computational interest, this advance has finally allowed the field of energetic pentazolates to become established and to flourish,with many energetic derivatives of the pentazolate anion now being known and fully characterized. The exceedingly high heat of formation of the pentazolates bodes well for their future inclusion in advanced energetic materials; however, derivatives with increased thermal stability will be needed before such inclusion is possible. The computational work that was done prior to experimental efforts has proven to be invaluable in guiding synthetic efforts and the development of new pentazolate-based energetic materials,thereby exemplifying the productive synergy that is possible between experimentalists and theoreticians.
Acknowledgements
We thank the grant from Army Research Office(ARO)(W911NF-18-1-0463),the Office of Naval Research(ONR)and Purdue University for the support of our lab.
Compliance with ethics guidelines
Dominique R. Wozniak and Davin G. Piercey declare that they have no conflict of interest or financial conflicts to disclose.
