
1例生物素酶缺乏症患儿的临床分析
2021-01-21 04:33吴镇宇罗建峰
中国妇幼健康研究 2021年1期
吴镇宇,罗建峰
(空军军医大学第一附属医院儿科,陕西 西安 710000)
生物素酶缺乏症(biotinidase deficiency)是由于生物素酶基因(biotinidase gene,BTD)突变引起生物素吸收和利用障碍的常染色体隐性遗传病,常表现为严重的神经、皮肤损害及代谢紊乱,病死率和致残率高[1];补充生物素治疗能起到良好的疗效,因此早期诊断和早期治疗是提高患儿生存率和生活质量的关键。本文报道1例以神经系统为主要表现的生物素酶缺乏症病例,对其临床表现、实验室检查、诊断及治疗等方面进行分析,以提高临床医生对该病的诊疗水平。
1资料与方法
1.1病史资料
患儿男,2个月21天,回族,因“间断抽搐1月余,咳嗽20余天,反应差3天”于2019年5月入住空军军医大学第一附属医院儿科。患儿生后27天无明显诱因出现抽搐,表现形式为意识丧失,双眼凝视,双手握拳,四肢僵直轻微抖动,伴口吐泡沫、口周发绀,每次持续数秒钟至1分钟不等,每天发作1~2次。曾于本院儿科住院治疗6天,期间查血乳酸偏高,为3.5mmol/L,血浆氨正常,尿三氯化铁实验阴性,行24小时视频脑电图显示醒-睡各期双侧额极、额、中央、颞区,出现较多尖波、多棘波、尖慢波同步或不同步发放,诊断为“癫痫、先天遗传代谢性疾病不除外”,予以“左乙拉西坦”口服抗癫痫治疗,半月余未再发作抽搐,并外送全外显子基因检测。本次入院20余天前,患儿出现咳嗽,呈阵发性连声咳,有痰不易咳出,抽搐再次发作,表现形式及持续时间大致同前,抽搐发作更加频繁,每天发作2~4次,曾于当地县级医院住院治疗,咳嗽减轻,仍频繁抽搐。近3天患儿出现反应及吃奶差,为进一步诊治来本院就诊。患儿系第4胎第4产,足月顺产,出生体质量为2.6kg,无窒息抢救病史。患儿精神运动发育水平落后于同龄儿,目前不能逗笑,不追视,不注视,不能抬头,不能抓拿东西。患儿无癫痫家族史,父母非近亲婚配,有3个姐姐,1个生后即出现抽搐,未诊治,3月龄夭折,其余2个姐姐体健。
1.2体格检查情况
患儿生命体征平稳,体质量5.7kg;反应差,嗜睡状态;阴囊、腹股沟区可见片状红色皮疹,头发细黄、稀疏,前囟1.5cm×1.5cm,平软,双侧瞳孔等大等圆约2mm,对光反射灵敏;咽部稍充血,双肺呼吸音粗,可闻及细湿啰音及少许喘鸣音;心、腹查体未见异常;四肢自主活动少、肌张力低下;双侧Babinski征阳性,双侧Chaddock征阳性。
1.3初步辅助检查情况
2结果
2.1患儿病情的发展及治疗
根据患儿病史及检查资料,临床诊断为癫痫、先天性遗传代谢病,合并感染、肝功能损害等;因外送血尿遗传代谢病筛查及全外显子基因测序结果未回报,初步予以左乙拉西坦、苯巴比妥钠抗癫痫,头孢噻肟舒巴坦钠控制感染,补液等治疗。患儿入院后病情进行性恶化,出现拒乳,不哭不闹,意识障碍逐渐加重,入院第2天复查血气分析:pH为7.31、PO2为114.0mmHg、PCO2为38.7mmHg、BE为-6.8、HCO3-为18.9mmol/L,显示严重代谢性酸中毒;予以纠正酸中毒及维持水电解质平衡等对症治疗,入院第3天复查血乳酸上升至9.1mmol/L,血气分析:pH为7.14、PO2为55.8mmHg、PCO2为82.1mmHg、BE为-3.7、HCO3-为27.6mmol/L,随后出现呼吸、心力衰竭,家长拒绝抢救及进一步治疗,患儿随后不幸夭折。
2.2特殊检查情况
患儿放弃治疗数天后,血串联质谱遗传代谢病检测显示:3-羟基异戊酰肉碱增高,伴丙酰肉碱增高,提示多种羧化酶缺乏;多种氨基酸增高,见图1、图2。尿有机酸气相色谱(GS/MS)检测显示:乳酸、2-羟基丁酸、3-羟基丙酸、丙酮酸、3-羟基丁酸、2-羟基异戊酸、3-羟基异戊酸、3-甲基巴豆酰甘氨酸及甲基枸橼酸增高;4-羟基苯乳酸增高,可继发肝功能损害,见图3。基因检测全外显子芯片捕获及高通量测序结果显示,BTD基因发现两处杂合突变(c.868G>A,p.G290R;c.38_44delGCGGCTGins TCC,p.C13fs),经家系验证构成复合杂合突变,该双杂合突变分别来自于父母,见图4、图5。
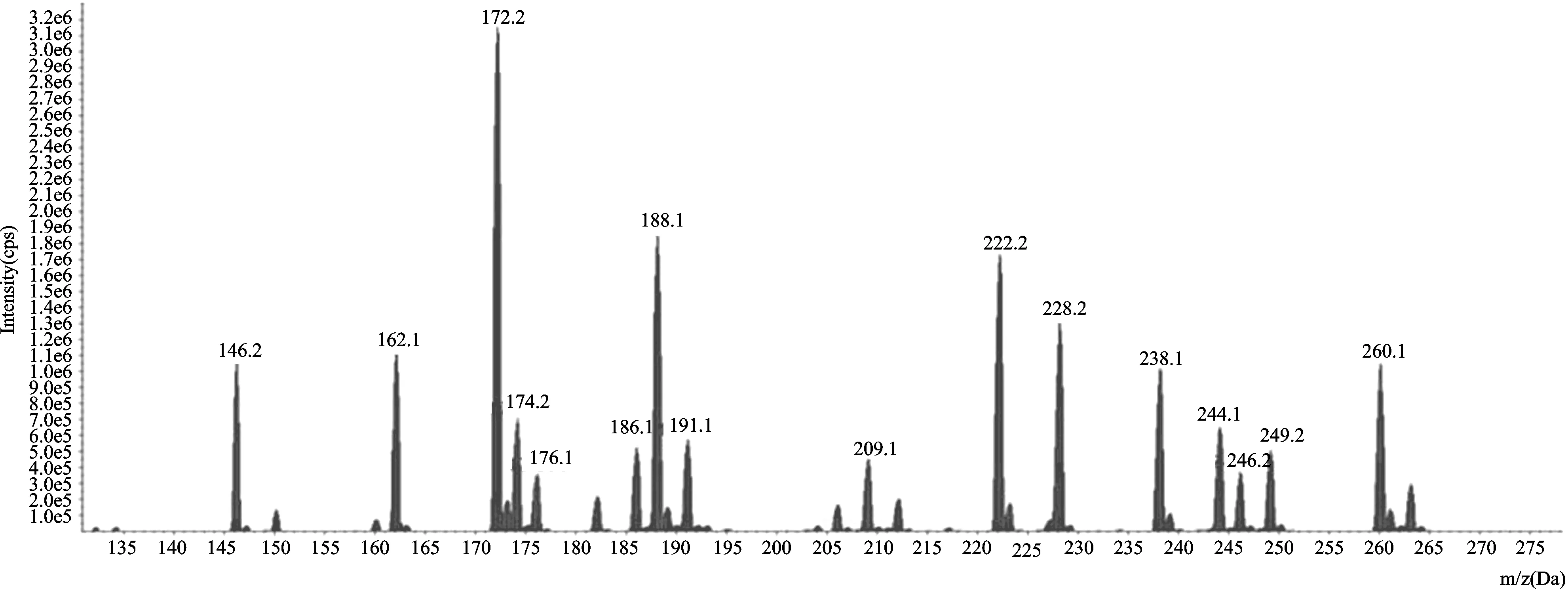
图1 氨基酸扫描图谱

图2 肉碱扫描图谱

图3 尿有机酸扫描图谱

图4 BTD,c.868G>A,染色体位置:chr3:15686291家系验证测序图
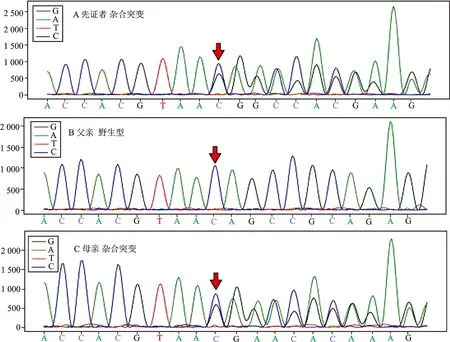
图5 BTD,c.38_44delGCGGCTGinsTCC,染色体位置:chr3:15676984家系验证测序图
3讨论
3.1生物素的生理功能
生物素(biotin)又称维生素B8、辅酶R,属于复合维生素B族,是一种人体不可缺少的水溶性含硫维生素。在机体的主要功能是作为线粒体内丙酰辅酶A羧化酶、丙酮酰羧化酶、乙酰辅酶A羧化酶、甲基巴豆酰辅酶A羧化酶等4种羧化酶的辅酶因子,通过羧化、脱羧和脱氢反应酶系参与三大营养物质的代谢;另外,生物素在调控基因表达中也起到重要作用[2]。生物素广泛分布于植物及微生物体内,但食物中的生物素呈蛋白结合状态,需要在肠道中生物素酶的作用下生成游离的生物素才能发挥生理作用[3]。
3.2生物素酶缺乏症的发病机理及临床表现
生物素酶缺乏症是由于BTD突变,引起生物素酶活性下降,人体吸收和利用生物素发生障碍,生物素依赖的多种羧化酶活性下降,线粒体能量合成障碍,不能够正常处理重要的代谢物质,引起代谢紊乱和一系列复杂多样的临床表现[4]。1983年Wolf等首次对生物素酶缺乏症进行了报告,随着遗传代谢病筛查技术水平的提高,该病在世界各国不断被报道。Lara等[5]对巴西一地区出生的新生儿进行该病的筛查结果显示,该地区的发病率为1/22 861,明显高于一些其它地区的报道。我国目前尚无该病流行病学的调查,只有数篇个案报道。生物素酶缺乏症多于婴幼儿时期起病,临床表现复杂,个体差异较大,全身各个系统均可受累,严重的神经系统损害更为明显,可表现为癫痫、发育落后、肌张力低下、痉挛性瘫痪、神经性耳聋和视神经萎缩等,其中癫痫为最常见的表现。杨艳玲等[6]对6例生物素酶缺乏症患儿进行了临床分析,其中3例表现为惊厥和智力运动落后,全部患儿均有运动障碍和肌张力异常,有5例经化验存在代谢性酸中毒。
本例患儿生后不足1个月即出现反复抽搐,伴精神运动发育迟滞,行脑电图检查显示多灶性放电及片段高峰失律,化验示血乳酸值偏高,结合患儿有1位姐姐发病与其相似,考虑患儿为先天遗传代谢性疾病继发癫痫,具体系何种遗传代谢病不能明确,外送基因检查,结果未回报前初步予以左乙拉西坦等抗癫痫治疗,效果差,抽搐发作未能控制。患儿再次入院前出现呼吸道感染,可能诱发了该病的急性发作,随后病情逐渐恶化,并出现严重代谢性酸中毒和高乳酸血症等代谢紊乱。
3.3生物素酶缺乏症的筛查和诊断
生物素酶缺乏症的筛查和确诊主要依靠血尿代谢病筛查、血生物素酶活性测定及基因检测[7]。典型患儿的血串联质谱分析显示3-羟基异戊酰肉碱、丙酰肉碱明显增高;尿有机酸分析显示乳酸、3-羟基丙酸、丙酮酸、3-羟基异戊酸、3-甲基巴豆酰甘氨酸及甲基枸橼酸等代谢产物增高,与该患儿血尿筛查结果相符。对患儿进行血生物素酶活性测定可见活性明显减低,低于正常均值的10%为完全性缺乏,介于正常均值的10%~30%为部分性缺乏[8]。
BTD基因位于3p25,全长约23kb,由4个外显子和3个内含子组成,目前已经发现多种突变可导致生物素酶缺乏症[9]。本资料在患儿BTD基因中发现c.868G>A和c.38_44delGCGGCTGinsTCC两处突变,一处为整码突变,另一处为移码突变,经家系验证,分别来源于父母,构成复合杂合突变,影响编码蛋白功能,符合常染色体隐性遗传规律。该病经早期口服生物素可以获得较好的治疗效果,推荐起始剂量为每天5~40mg,并根据不同个体的临床表现及实验室检查结果调整用药剂量,一般数日后尿异常代谢产物消失,全身症状能够显著改善。王红梅等[10]对14例给予生物素治疗的生物素酶缺乏症患儿进行了4个月至2年的随访发现,患儿的症状均全部明显改善,复查生物素水平均增高。遗憾的是本资料中患儿明确诊断前已夭折,未进行补充生物素治疗,如果提前给予治疗,预后可能良好。
综上所述,生物素酶缺乏症患儿临床表现复杂,常导致严重神经系统损害,临床诊断困难,对婴幼儿期发病的癫痫伴发育落后患儿需考虑生物素酶缺乏症的可能,因此,应及早行血尿代谢病筛查及基因检测,确诊后及早予以补充生物素治疗。
猜你喜欢
今日农业(2021年16期)2021-11-26
现代畜牧科技(2021年6期)2021-07-16
现代畜牧科技(2021年6期)2021-07-16
兽医导刊(2020年23期)2020-12-30
中国畜牧杂志(2019年4期)2019-04-20
猪业科学(2018年5期)2018-07-17
农村青少年科学探究(2017年6期)2017-09-11
家庭百事通·健康一点通(2017年3期)2017-03-22
现代检验医学杂志(2016年4期)2016-11-15
合成化学(2015年4期)2016-01-17
