
微小RNA在COPD发病机制中的研究进展
2021-01-19 06:17:58廖志文周建荣
赣南医学院学报 2020年11期
廖志文,周建荣
(1. 赣南医学院2018级硕士研究生;2. 赣南医学院第一附属医院呼吸内科,江西 赣州 341000)
慢性阻塞性肺疾病(Chronic obstructive pulmo⁃nary disease,COPD)是目前全球最常见的慢性疾病之一,2018 年我国COPD 患病人数超过了1 亿,是导致卫生资源负担增加和生活质量降低的重要原因[1-2]。环境与遗传的相互作用是COPD发生的关键过程,而炎症、氧化应激、蛋白酶与抗蛋白酶失衡被认为是重要的病理特征[3]。以往发现的重要环境因素如吸烟、职业环境暴露、大气污染,能诱导肺结构细胞和免疫细胞功能异常[4],这些异常的细胞共同参与了COPD的病理形成[5]。
微小RNA(MicroRNA,miRNA)是一种长度为18~24 个核苷酸的非编码RNA(Non-coding RNA,ncRNA),通过与mRNA的3′非翻译区(3′untranslated region,3′UTR)结合,在转录后抑制靶mRNA 表达或促进mRNA 降解[6]。miRNA 种类丰富,每种miRNA可与多个不同的靶mRNA 结合,被认为控制着人类30%~60%的基因的表达,参与细胞增殖、分化、凋亡、癌变和免疫反应[7-8]。miRNA 可被外部环境诱导表达改变,这些miRNA 的改变引发肺结构细胞和免疫细胞功能失调,促成COPD 的病理改变。本文就miRNA在与COPD发病相关的细胞中的作用机制最新研究进展进行综述。
1 MiRNA与气道上皮细胞
气道上皮细胞作为肺的保护屏障,通过粘液纤毛清除机制清除微生物和外来颗粒[9]。烟雾(Cigarette smoke,CS)或其他吸入刺激物可诱导气道炎症反应和上皮细胞的衰老、凋亡[4]。长期暴露于这些刺激下的人群和COPD 患者中肺内和血液中多种miRNA 表达水平发生改变,一些miRNA 被发现参与气道上皮细胞功能调控,如miR-29b 参与炎症因子的调控;miR-218、miR-146a、miR-181a-2-3p 调节炎症激活通路;miR-126、miR-570-3p 与细胞衰老有关;miR-21、miR-145-5p 参与细胞凋亡。TANG等[10]研究发现,miR-29b 在吸烟者的肺组织、血浆中表达水平下调,而在COPD 患者中进一步降低,且与肺功能指标、白介素(Interleukin,IL)-8 水平相关。荧光素酶报告分析显示,转录共调控因子BRD4(Bromodomain protein 4)基因是miR-29b 的调控靶点。体外香烟烟雾提取物(Cigarette smoke extract,CSE)处理的人支气管上皮细胞(Human bronchial epithelial cells,HBEC)中miR-29b 同样出现下调,导致BRD4 表达增加,进而增强下游的IL-8 基因转录。另外,抗氧化剂能防止CSE 诱导的这些改变,表明CS 可以通过氧化应激来诱导miRNA 表达改变。氧化应激直接损伤肺上皮细胞,同时引起DNA 损伤和细胞功能受损,加重细胞衰老、凋亡,增强炎症反应[11]。PASCHALAKI 等[12]发现,与吸烟有关的miR-126 在COPD 患者的肺上皮细胞中表达降低,并在CS 暴露后的小鼠模型实验中证明,下调的miR-126可以增强激活DNA 损伤通路的相关酶ATM(Ataxia telangiectasia mutated)蛋白激酶的活化,促进HBEC的衰老和功能障碍。Sirtuin-1 是一种衰老和炎症的 重 要 调 节 因 子,与COPD 细 胞 衰 老 有 关[13]。miR-570-3p 在COPD 肺组织中表达增加,体外实验显示,miR-570-3p 同样也可被氧化应激诱导表达上调,并通过直接抑制HBEC中sirtuin-1基因表达促进细胞衰老,增强炎症因子的分泌,而miR-570-3p 拮抗剂能逆转这些改变[14]。miR-570-3p 可能成为衰老治疗的重要靶点。气道上皮细胞增殖减缓和凋亡增加也是COPD 气道中的重要特征,miR-21 被发现在CS 诱导的COPD 小鼠肺组织和CSE 处理后的HBEC 中表达上调,并参与了CS 诱导的HBEC 自噬和凋亡[15]。DANG等[16]发现吸烟者、COPD患者肺组织中miR-145-5p 的表达下调,进一步的体外实验证实,miR-145-5p 可直接抑制介导细胞凋亡的相关基因KLF5(Kruppel-like 5)的表达,减轻CS 诱导的HBEC 凋亡和炎症反应。然而,DUTTA 等[17]发现,miR-145-5p 在暴露于CS 后的小鼠肺组织中表达是上调的,并进一步证明,CS 通过转化生长因子β(Transforming growth factorβ,TGF-β)诱导HBEC 中miR-145-5p表达的上调,并导致气道粘液清除功能受损。这与DANG 等[16]研究中COPD 患者miR-145-5p表达下调的结果相反,两者研究结果的不同在很大程度上取决于CS 暴露时间和强度。COPD 的发生是长期接受外界刺激的过程,这些miRNA 在随病程延长而发生的改变可能反应了肺的代偿机制。miRNA 在人类基因的调控网络十分复杂,可能同时存在多种其他调控途径。最近,SONG 等[18]在体外实验利用双荧光素酶报告分析证实,长ncRNA(Long noncoding RNA,lncRNA)母体表达基因3(Maternally expressed gene 3,MEG3)能与miR-218结合并调控miR-218 的表达,并共同参与CSE 诱导的网络在调控气道上皮细胞的功能上也具有重要的作用。在之前的研究中发现,miR-218 在COPD患者血清中表达下调,并且与吸烟有关,进一步的体外荧光素酶报告分析证实,HBEC 中可溶性肿瘤坏死因子受体(Tumor necrosis factor receptor,TNFR)1 的3′UTR 是miR-218 直 接结合靶点,miR-218 通过抑制HBEC 中TNFR1/NF-κB(NF-kappa B)信号的激活抑制CSE诱导的炎症反应和粘蛋白MUC5AC高分泌[19]。miRNA 可能是气道上皮抵御CS 诱导的炎症激活的重要调节因子,而这些过程主要是通过调节NF-κB信号通路介导的。miR-146a、miR-181a-2-3p也被发现参与HBEC 中NF-κB 信号通路的调控,两者分别被认为在亚微米颗粒物和金属镉诱导的炎症反应中充当负反馈调节因子。LIU 等[20]在体外实验发现,HBEC 中miR-146a被亚微米颗粒物通过激活NF-κB 信号通路诱导表达上调,并直接抑制NF-κB信号通路上游基因表达。KIM 等[21]利用全基因表达谱筛选出两个与NF-κB 激活相关的基因,Toll 样受体(Toll-like receptor,TLR)4和SQSTM1/p62基因,被认为可能是miR-181a-2-3p潜在调控靶点。然而,与miR-181a-2-3p 不 同 的 是,miR-146a 无论 在COPD 患者还是体外亚微米颗粒物诱导的HBEC中表达均为上调[22],miR-146a 的表达上调对COPD的炎症的负反馈作用似乎是有利的,并且这种作用持续存在COPD 的发病过程中,而miR-181a-2-3p 表达被诱导下调,导致炎症激活的相关基因表达增强,持续和不可逆的炎症激活最终导致了COPD 持续的慢性炎症反应。这表明一些具有类似生理功能的miRNA 被外界诱导表达改变时,造成的结果并不是都是同向的,而这种有利的miRNA 变化可能随病程延长不足以抵抗失衡的miRNA 引起的不利改变,最终导致了不可逆的病理改变,这些过程可能反映了体内miRNA网络的代偿机制。
2 MiRNA与肺成纤维细胞
肺成纤维细胞(Lung fibroblasts,LF)主要作用是合成和维持细胞外基质,被认为是维持肺泡结构完整性的关键。CS 能干扰LF 的增殖和修复作用,抑制细胞外胶原凝胶收缩,而LF功能障碍是肺气肿和小气道纤维化发生的重要原因[23-24]。CS 诱导LF中的miRNA 失调,这些miRNA 参与LF的增殖、修复和细胞因子分泌调控等。ONG 等[25]通过在正常人的肺组织和LF 中进行RNA 测序发现,当前吸烟人群中miR-335-5p 的表达较过去吸烟和从不吸烟人群明显下调,并用RT-qPCR 方法再次得到验证,而3个与细胞增殖有关的基因,RB1、CARF和SGK3通过Argonaute 2 免疫沉淀法被鉴定是LF 中miR-335-5p的靶点。TGF-β 被认为是LF 增殖和修复功能重要调节因子。同一团队[26]通过对TGF-β 刺激和未刺激的COPD 患者、健康者LF 中进行RNA 测序,发现了多种TGF-β 调节和与COPD 相关的miRNA,其中COPD 患者中miR-660-5p 较健康者表达显著上调,miR-27a-5p经TGF-β诱导表达显著上调,miR-21-5p、miR-148b-3p、miR-589-5p 和miR-376b-3p 在COPD患者中对TGF-β 的反应较健康者减弱,进一步用上述相同方法预测了miR-27a-5p、miR-148b-3p 和miR-660-5p 的多个靶点存在于与基因转录调控和细胞增殖相关的基因。TGF-β 介导调节的miRNA可能对LF功能调控具有重要作用,而TGF-β的异常释放和LF 对TGF-β 刺激反应改变可能是造成COPD中LF功能障碍的重要机制。
低氧诱导因子1α(Hypoxia-inducible factor 1α,HIF-1α)是调节缺氧反应的重要转录因子,在COPD的发生发展中发挥着重要作用。高表达的HIF-1α能诱导肺细胞凋亡,激活表皮生长因子受体/磷脂酰肌醇激酶/AKT 通路,上调炎症因子的表达[27]。LIN等[28]发 现,miR-186 转染后的LF 增殖 明 显 减少,Western blot 分析结果显示HIF-1α 蛋白水平降低,但仍高于阴性对照组水平,双荧光素酶报告分析提示HIF-1α mRNA 的3′UTR 存在miR-186 的结合位点,这表明miR-186 可能调节HIF-1α 表达影响炎症水平和LF 增殖功能,然而,miR-186 抑制剂转染后的LF 增殖和HIF-1α 表达水平变化并不明显,这可能是由于miR-186 在正常的LF 中的表达处于较低水平。LF 修复功能受多种方式调节,包括生长因子的释放。血管内皮生长因子(Vascular endothelial growth factor,VEGF)是一种有效的血管生成和血管通透性调节因子,由HIF-1α 诱导表达,在COPD 的组织重塑和血管生成中发挥作用[29]。miR-503 在COPD 患者的LF 中的表达降低,体外实验证实,miR-503 通过直接抑制VEGF mRNA 的表达,调节LF 的VEGF 的生成[30]。炎症的限速酶环氧化酶-2(Cyclooxygenases-2,COX-2)是在miR-146a的调控靶点已被证实。ZHOU 等[31]研究发现,lncRNA PVT1可以诱导LF 中miR-146a 启动子的甲基化,影响的miR-146a 加工和成熟,进而调控下游COX-2 基因的表达。另外,miR-146a 还参与HBEC 中炎症反应的负反馈调节[20]。OSEI 等[32]发现,气道上皮细胞来源的IL-1α增加LF中miR-146a-5p的表达,miR-146a-5p同时通过下调LF 中IL-1 受体相关激酶表达对气道上皮源性的IL-1α 诱导的IL-8 分泌产生负调控作用,而COPD 患者LF 中miR-146a-5p 对IL-1α 的反应减弱而表达增加减少,IL-8 分泌受到的抑制作用减弱。
3 MiRNA与免疫细胞
COPD 的炎症反应是多种细胞及炎症因子参与的过程,除结构细胞外,参与的免疫细胞包括肺泡巨噬细胞(Alveolar macrophages,AM)、中性粒细胞、淋巴细胞、嗜酸性粒细胞等,这些被激活的免疫细胞分泌多种炎症介质、抗体、蛋白酶等对肺组织细胞产生细胞毒性;这种炎症过程在随后的长期的环境刺激下被持续激活和放大,并对COPD 的病理改变起着关键作用[5]。AM 位于肺上皮表面,经氧化应激激活后分泌多种趋化因子和细胞因子,在COPD的慢性炎症中发挥关键的协调作用[5,33]。吸烟可以改变AM 中多种miRNA 的表达,其中一些miRNA 参与调节细胞分泌水平,如let-7c 通过抑制转录激活因子(Signal transducer and activator of transcription,STAT)3 的表达调节AM 炎症因子释放[34]。近期的研究发现AM 的激活和极化也与miRNA 有关,而NF-κB 相关通路中的基因常是其调控靶点。WANG等[35]发现miR-27-3p 在动物模型中表达上调,并进一步证明,miR-27-3p 通过抑制过氧化物酶体增殖物激活受体γ(Peroxisome proliferator-acfivated recep⁃torγ,PPARγ)的表达以增强AM 中的TLR2/4 信号传导,进一步介导TLR2/4 信号下游的NF-κB、JNK/p38、JAK/STAT 通路,增强CS 和脂多糖诱导的炎症反应,同时促进AM活化,影响AM的极化。miR-223也通过调控NF-κB 信号通路激活上皮细胞的炎症反应,最近的研究还证明,miR-223 是中性粒细胞募集的重要调节因子[36]。中性粒细胞在COPD 患者的气道中被募集和异常活化,并与疾病严重程度相关。这表明miR-223 在COPD 中的炎症细胞网络中可能充当着重要的角色。T 淋巴细胞在COPD 患者肺实质及气道数目增加,与肺泡细胞破坏和凋亡密切相关[5]。DIAO 等[37]发现miR-132 在COPD 患者和吸烟者血清中的表达上调,与CD8+T 细胞数目比例相关,体外荧光素酶报告分析显示,miR-132 可靶向结合转导抑制因子5(Suppressor of cytokine signal⁃ing 5,SOCS5)mRNA 的3′UTR,而SOCS5 是淋巴细胞的发育和功能的重要调节因子,特别是CD4+T 细胞。SOCS5 基因表达减少可能引起CD4+T 数目的增殖减少,导致杀伤性CD8+T 细胞比例数上升,这可能是COPD 中肺泡细胞破坏和凋亡增加的重要原因。WEN 等[38]研究发现在COPD 患者和吸烟者外周血中miR-3202 表达下调,进一步的体外实验显示,CSE 诱导T 淋巴细胞中miR-3202 水平下调,共培养的HBEC 凋亡增加,而miR-3202 转染的T 淋巴细胞可减轻这种反应。双荧光素酶法证实,miR-3202 能通过直接抑制T 淋巴细胞中FAS 凋亡抑制分子2 基因的表达减弱T淋巴细胞的杀伤作用。
4 MiRNA与细胞交流
各种肺结构细胞群和免疫细胞亚群的协调功能构成了一个复杂的细胞间通讯网络,这对肺部稳态和修复至关重要[39]。大多数前期关于COPD 中miRNA 的功能和作用的研究局限于特异种类的细胞内,而细胞间的联系很少被提及(表1)。上述提及的miR-146a、miR-223分别被认为可能是COPD中异常HBEC 与LF、HBEC 与免疫细胞间串音的关键介质,这表明细胞外的miRNA 可能作为一种细胞通讯介质介导细胞网络功能调控。细胞外囊泡(Extra⁃cellular vesicle,EV)是细胞旁分泌产生的膜性囊泡而存在于多种体液中,包括凋亡小体、微囊泡和外泌体,其作为细胞通讯的重要载体可以传递蛋白质、ncRNA、脂质、mRNA、DNA 等[40]。吸烟者和COPD 患者中多种EV-miRNA 的表达水平发生改变[41]。最近的研究证明,EV-miRNA 参与COPD 的发生和进展,并已成为当前的研究热点。miRNA 的异常传递可能促进了原细胞中miRNA 的失调,而这种传递可发生同种或不同种细胞间,最终导致整条细胞网络功能异常。GUPTA 等[42]研究发现,在气道上皮细胞之间的EV-miRNA 和EV-粘蛋白MUC5B传递可以增加气道分泌,促进气道重塑的发生。HBEC-LF 异常通讯也是气道重塑的重要因素,而miR-21 在上述HBEC、LF 中参与气道修复和重塑过程[15,26]。XU 等[43]通过体外实验发现,HBEC 来源的外泌体miR-21 可被CSE 诱导转移到LF 中,并通过pVHL(Von Hippel-Lindau protein)/HIF-1α 信号通路促成肌成纤维细胞分化表型,同样在CS诱导的小鼠模型上也得到验证。另外,免疫细胞与肺上皮细胞间也可发生EV-miRNA 转移。miR-223 可以经巨噬细胞或单核细胞微泡的分泌输送到内皮细胞等非免疫细胞中[36],可能充当肺部炎症反应中重要的细胞间通讯介质。中性粒细胞胞外陷阱(Neutrophil extracellular traps,NETs)是由DNA 纤维和抗菌蛋白组成的细胞外网状结构,不仅可抵御病原体侵入,同时也可介导宿主免疫损伤,与多种炎性疾病有关,包括COPD、哮喘等[44]。NETs 可以调节巨噬细胞和HBEC的炎性因子分泌,但对于这些过程机制目前尚不清楚[45]。最近有研究表明,NETs可能是细胞外miRNA的一种新的载体。LINHARESLACERDA等[46]研究发现,NETs 能携带miRNA-142-3p,并可以转移到巨噬细胞中,调节TNF-α的产生。这表明NETs可能是通过携带miRNA 转移到巨噬细胞和HBEC 中调控炎症反应,但目前仍需要更多的研究来证实。
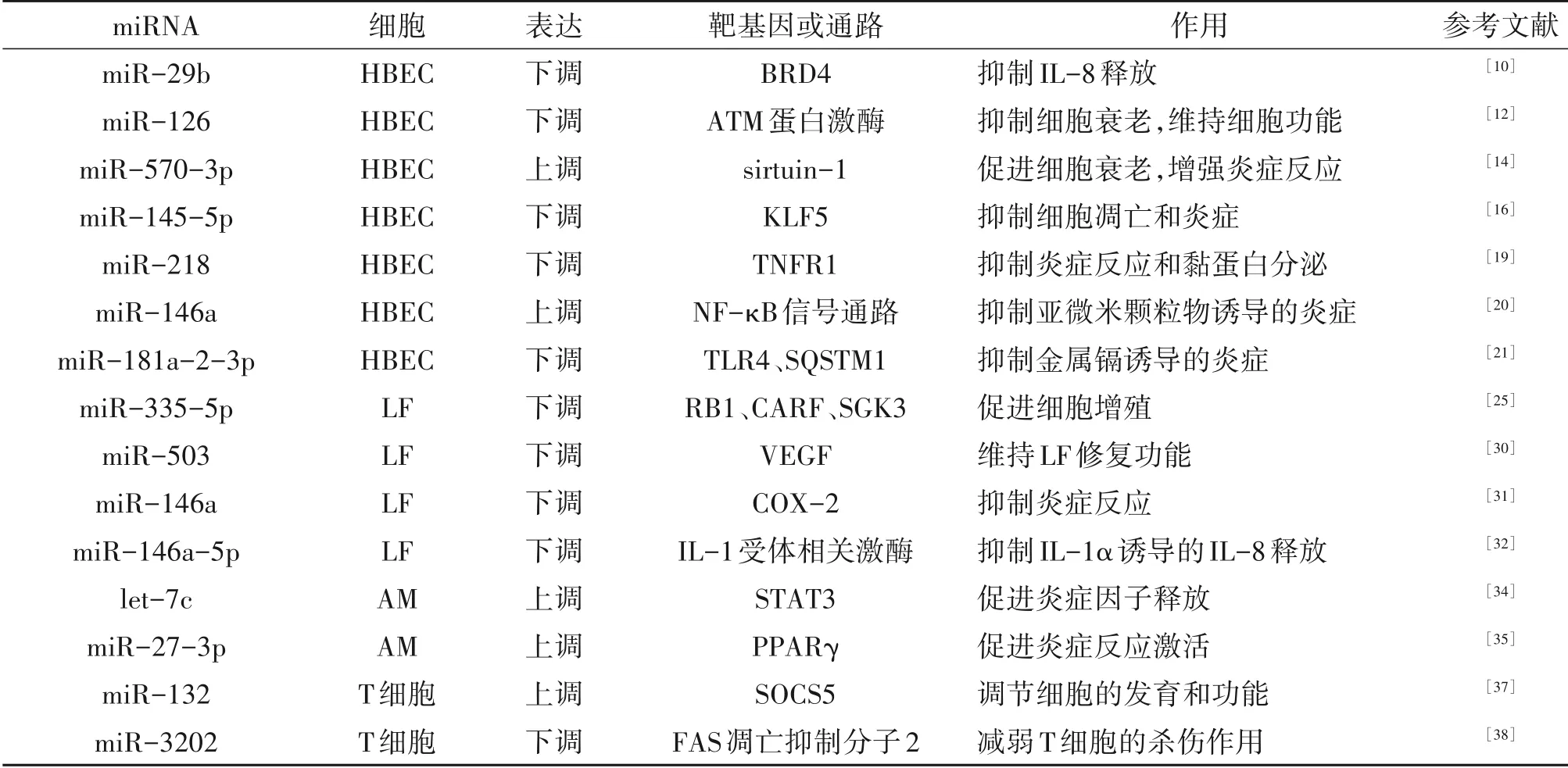
表1 COPD中差异性表达的miRNA的靶细胞、靶点及作用
5 总结和展望
COPD 的发生是环境和遗传共同的结果,涉及多种细胞和细胞因子参与。近年来,大量miRNA 在这些细胞中作用机制被逐渐阐明,将进一步从环境与遗传层面深入认识COPD的发生过程。miRNA作为COPD 生物标志物和治疗靶点的潜力也越发突显。但是,目前miRNA 在COPD 发病机制中的认识仍存在一定的局限性,miRNA 本身功能的高特异性和靶点的多样性是重要原因。随着miRNA 在参与COPD 发病的细胞内机制研究的不断深入,以及细胞外miRNA 的联系,特别是近年来EV-miRNA 与COPD 的研究,将更加全面的揭示COPD 发病中miRNA 的表观调控机制,并且EV 作为miRNA 的载体可能为当前COPD 的miRNA 靶向治疗研究提供一种更加精准的方式。
猜你喜欢
中老年保健(2021年3期)2021-12-03 02:32:25
中国生殖健康(2020年7期)2020-12-10 07:48:51
学苑创造·A版(2020年12期)2020-01-07 14:07:23
国际呼吸杂志(2019年22期)2019-12-09 09:20:26
中国外汇(2019年15期)2019-10-14 01:00:34
国际呼吸杂志(2019年5期)2019-03-30 01:38:20
国际呼吸杂志(2019年3期)2019-03-01 05:39:06
国际呼吸杂志(2019年2期)2019-02-14 06:11:26
作文教学研究(2016年1期)2016-07-05 12:22:47
医学研究杂志(2015年8期)2015-06-22 14:00:57
