
Identification of Potential Therapeutic Targets of Alzheimer’s Disease By Weighted Gene Co-Expression Network Analysis
2021-01-09 03:38FanZhangSiranZhongSimanYangYutingWeiJingjingWangJinlanHuangDengpanWuZhenguoZhong
Chinese Medical Sciences Journal 2020年4期
Fan Zhang,Siran Zhong,Siman Yang,Yuting Wei,Jingjing Wang,Jinlan Huang,Dengpan Wu,Zhenguo Zhong*
1Pharmacy School,Guangxi University of Chinese Medicine,Nanning 530200,China
2Jiangsu Key Laboratory of New Drug Research and Clinical Pharmacy,Pharmacy School,Xuzhou Medical University,Xuzhou,Jiangsu 221004,China
Key words:bioinformatics analysis; Alzheimer’s disease; Tricarboxylic acid (TCA) cycle;weighted gene co-expression network analysis; OXCT1; ATP6V1A
Objective Alzheimer’s disease (AD) is the most common cause of dementia.The pathophysiology of the disease mostly remains unearthed,thereby challenging drug development for AD.This study aims to screen high throughput gene expression data using weighted co-expression network analysis (WGCNA) to explore the potential therapeutic targets.Methods The dataset of GSE36980 was obtained from the Gene Expression Omnibus (GEO) database.Normalization,quality control,filtration,and soft-threshold calculation were carried out before clustering the co-expressed genes into different modules.Furthermore,the correlation coefficients between the modules and clinical traits were computed to identify the key modules.Gene ontology and pathway enrichment analyses were performed on the key module genes.The STRING database was used to construct the protein-protein interaction(PPI) networks,which were further analyzed by Cytoscape app (MCODE).Finally,validation of hub genes was conducted by external GEO datasets of GSE 1297 and GSE 28146.Results Co-expressed genes were clustered into 27 modules,among which 6 modules were identified as the key module relating to AD occurrence.These key modules are primarily involved in chemical synaptic transmission (GO:0007268),the tricarboxylic acid (TCA) cycle and respiratory electron transport (R-HSA-1428517).WDR47,OXCT1,C3orf14,ATP6V1A,SLC25A14,NAPB were found as the hub genes and their expression were validated by external datasets.Conclusions Through modules co-expression network analyses and PPI network analyses,we identified the hub genes of AD,including WDR47,OXCT1,C3orf14,ATP6V1A,SLC25A14 and NAPB.Among them,three hub genes (ATP6V1A,SLC25A14,OXCT1) might contribute to AD pathogenesis through pathway of TCA cycle.
ALZHEIMER’S disease (AD) is the most common cause of dementia,leading to impaired cognition,memory,language,and dysfunctions in daily activities.The pathological characterization includes formation of neurofibrillary tangles and amyloid plaques in brain.[1,2]Familial AD,linked with APP or PS1 mutation,involves less than 1% of the AD population.The sporadic AD accounts for over 99% of the cases,although the etiology largely remains unknown.Moreover,based on the current theories,no therapy can effectively prevent the occurrences of AD.[3]Therefore,the exact underlying mechanism of the onset of AD and its molecular basis remain to be fully elucidated.
Weighted gene co-expression network analysis(WGCNA)[4]is a systematic biology procedure to explore clusters (modules) of highly correlated genes between different groups of samples,and to analyze the association of these modules with clinical traits to illustrate the trait-related key modules.Lastly,intramodular analysis and network visualization could be performed to identify the key genes within modules,which could further facilitate the discovery of novel therapeutic targets or candidate biomarkers.
In this study,we conducted WGCNA on GSE36980 of AD and normal controls.In each group,three tissue samples of different parts of brain were studied.Initially,27 modules were elucidated by constructing the weighted co-expression network.Computation of the correlation coefficients between the modules and clinical traits,six key modules were identified.Intramodular analysis was executed; gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were then conducted.After network hub genes screening and hub genes validation,we determined three hub genes (ATP6V1A,SLC25A14,OXCT1)that may be involved in AD by regulating tricarboxylic acid (TCA) cycle,and these hub genes may be explored as the potential targets for AD therapy.
MATERIALS AND METHODS
Data source
Based on the platform of GPL6244 Affymetrix Human Gene 1.0 ST Array,the mRNA microarray dataset of GSE36980 and corresponding metadata were downloaded from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/).GSE36980 included three tissue samples of frontal cortex,hippocampus,and temporal cortex from postmortem brains(Homo sapiens) of overall 32 AD donor and 47 normal controls.
WGCNA analysis
R software (version 3.6.1) with WGCNA 1.67 package[4]was utilized to process the dataset.Dataset normalization was performed by limma 3.38.3 package[5]and then verified for outliners.T-distributed stochastic neighbor embedding (T-sne) and principal components analysis (PCA) were employed for quality control.Thereafter,the probes,with median expression less than 1/5 of the average level,were screened out.The standard deviation (SD) of the remaining gene expression was computed.For further analysis,the top 10,000 most variant genes were selected.To achieve a reasonable scale-free topology fit,the soft-thresholding power[6,7]was calculated initially and the adjacencies were evaluated.Next,the adjacency was transformed into Topological Overlap Matrix (TOM) to minimize the effects of noise.Finally,a dendrogram was constructed by hierarchical clustering,and Dynamic Tree Cut[8]was performed to detect highly co-expressed genes (modules) according to their similarity in the TOM.
Identification of significant modules of clinical traits
Identification of the module-trait relationships was achieved by estimating the correlation between modules and clinical traits.Gene significance (GS) was analyzed to assess the correlation between module genes(eigengene) and clinical traits.Furthermore,the significant modules were located.The principal components of module gene expression were considered to be module membership (MM),which could measure the correlation of the module eigengene and the gene expression profiles.The genes with high GS and MM were investigated to identify the significant gene relevant to development of AD.Module gene with GS and MM higher than 0.5 or lower than–0.5 were identified as the hub gene.
Intramodular and intermodular analysis
Intramodular analysis[8]was performed on selected modules to explore the module biological functions,including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis,by Metascape[9]online database.GS and MM analyses within module were carried out to identify hub genes with high GS and MM.Correlation of eigengene was examined to reveal the intermodular relationship.
Network construction
Network constructions were conducted by calculating the interconnection weight of paired genes in key modules in TOM.Module genes with weight above 0.08 were selected and visualized by Cytoscape (version 3.7.0).[10]
Protein-protein interaction network construction
Genes in key modules were selected for Protein-protein interaction (PPI) network analysis by using the STRING database 11.0 (https://string-db.org/)[11]and Cytoscape.To isolate clusters with high interconnected regions or protein complexes in the network,the Cytoscape app MCODE 1.5 (http://apps.cytoscape.org/apps/mcode) was used.
Hub genes validations
Two GEO datasets were downloaded from the GEO database for hub genes validations.The microarray dataset of GSE1297 contained hippocampus tissues from 9 normal controls and 22 AD patients,as well as the corresponding clinical information including MiniMental Status Examination (MMSE) score.The microarray dataset of GSE28146 contained hippocampus tissues from 8 normal controls and 22 AD patients who were classified as three stages:incipient,moderate,and severe.To further explore the potential role of hub genes in cognitive and clinical features,pearson correlations between hub genes expression and MMSE score were calculated.
Statistical analyses
Intramodular and intermodular analyses were performed according to pearson correlation among different modules using WGCNA 1.67 package;[4]Pearson correlation,one-way analysis of variance (ANOVA)ort-tests were performed to validate the hub gene among AD patients with different stage and controls in GSE1297 and GSE28146 datasets by using SPSS 20.0 software.
RESULTS
Key modules of Alzheimer’s disease
After the normalization,quality control (supplementaryFigure S1),and filtration,the top 10,000 genes with the highest SD (standard deviation) were selected for WGCNA.We chose 7 as the soft-thresholding power by PickSoftThreshold function calculation (Figure 1A,1B).The dendrogram was constructed by setting minimum module size as 30 and Dynamic Tree Cut high as 0.25.There were 27 corresponding modules clustered(Figure 1C,Table 1).The genes which could not be attributed to any modules were categorized into the grey module.
Identification of the key modules and the key trait of AD
By the module-trait relationship heatmap based on the correlation between the modules and clinical traits(Figure 2),we identified 6 modules (darkturquoise,tan,black,salmon,grey60,and turquoise) as the key modules.According to the heatmap,the hippocampus had the highest correlation coefficient among 6 modules,indicating that the greatest alterations of gene expression between AD and controls were obtained in sample of hippocampus.Hence,the hippocampus was selected as a key trait of AD for further analyses.
Intramodular analyses and intermodular analyses
Estimation of the hub genes with a high correlation to AD status was achieved by conducting GS and MM analyses.To explore the differential expression of the hippocampus between AD and controls,we screened the 6 modules (turquoise,darkturquoise,salmon,tan,black,and grey 60).In each module,module hub genes were screened by correlation coefficient (GS and MM) for further analyses (Figure 3,A-F;supplementaryFigure S2),which provide unique insights into the biological mechanism of AD.Dendrogram of the eigengenes and heatmap (Figure 4) revealed high correlations of 3 modules (darkturquoise,tan,black) with the AD status,suggesting those module genes may exert relevant biological functions.On the other hand,3 modules (yellow,green,and blue) were also selected for GS and MM significant genes screening to explore differentially expressed genes among different tissues.Figure 3 G-Idemonstrated high correlation in those modules,which indicate hug amount of genes were differentially expressed among three tissues,suggesting they may play various roles in AD pathogenesis.
Module genes GO and KEGG analysis
Genes in black,tan,and darkturquoise modules were subjected to GO functional and KEGG pathway enrichment analyses by using online database of Metascape,and the results were shown inFigure 5.Tan and darkturquoise modules were primarily associated with synaptic functions,including chemical synaptic transmission(GO:0007268) and transmission across chemical synapses (R-HSA-112315).Meanwhile,the black module had significant association with citric acid (TCA) cycle and respiratory electron transport (R-HSA-1428517),signaling by ROBO receptors (R-HSA-376176).
Identification of hub genes
Network analyses on darkturquoise,tan,and black modules selected the top 36 genes,and the hub genes in networks were further screened out by degree and visualized.PCMT1,WDR47,OXCT1,C3orf14,ATP6V1A,SLC25A14,andNAPBwere screened out in darkturquoise,tan,and black modules (Figure 6A).AGAP6,AGAP7P,ATXN7in the grey60 module,MIR326,KRTAP5-3,RNA5SP408,RNA5SP413in thesalmon module,andRBFOX1,FRMPD4,DNM1,andLNX1in the turquoise module were identified as hub genes(Figure 6,B-D).
PPI network analysis
For the genes in darkturquoise,tan,and black modules,the PPI network constructed by the STRING database were shown inFigure 7.The network MCODE scores of Darkturquoise,tan,and black modules were measured,and visualized by color scaling.SLC32A1,GAD1,etc.,in the darkturquoise PPI network,EPHA7andTCEB1 in the tan PPI network,andRCHY1,TCEB1,FBXO4,etc.,in the black module PPI network showed high MCODE score and were screened out by PPI network nodes connectivity.Ten hub genes in the black module was identified by MCODE.The results of GO and KEGG enrichment analyses were presented inTable 2,which demonstrated a significant correlation of the key sub-network with the Class I major histocompatibility complex (MHC) mediated antigen processing (R-HSA-983169).
Hub genes validations
Expression of the hub gene in GSE 1297 and GSE 28146 datasets showed thatWDR47,OXCT1,C3orf14,ATP6V1A,SLC25A14,NAPB,RCHY1,EPHA7,and TCEB1 expressions were significantly different between controls and severe state AD patients,signifying their potential target roles in AD pathogenesis (Figure 8).The expressions ofOXCT1(r=0.547,P<0.01),C3orf14(r=0.564,P<0.001) were significantly correlated to the MMSE scores.However,how these genes participate in AD pathogenesis have so far not been investigated.
DISCUSSION
AD is a complex chronic neurodegenerative disease,which is characterized by decline in memory,language,motivation,and problem-solving ability.Moreover,significant progress has been achieved in the classical biomedicine based on molecular biology,which merely focus on individual genes and proteins that forms the basis of experimental biology.Since the pathophysiology of this complex disease remains unknown,it may be achieved by the breakdown of a significant amount of correlated genes rather than individual genes.Therefore,the application of network-based analysis[12]approaches could help to elucidate the dynamics of AD progression[13]or the expression patterns across different regions of the brain.[14]This study aims to understand the information captured by high-throughput experiments data which is far richer than a list of differentially expressed genes.
In this study,we conducted WGCNA on microarray datasets downloaded from the GEO database which contained the frontal cortex,hippocampus,and temporal cortex from AD patients and non-AD donors.The GSE36980 samples expression was heterogeneous between the 3 tissues and homogeneous between AD patients and non-AD patients (Figure 1).After normalization and filtering of the data,the top 10,000 genes with the highest SD were selected to prevent high correlation produced by genes without notable variance.Following gene co-expression network construction,27 modules were clustered and identified.
In module-trait relationship heatmap,the first column of trait illustrated significant correlation of 3 modules (darkturquoise,tan,and black) with AD.By comparing the three tissue traits separately,the probable contribution of hippocampus genes expression pattern to the onset of AD was identified.Furthermore,the salmon module exhibited a high correlation solely with Hippocampus.
PCMT1was identified as a hub gene through network visualization in the current study.A previous study[15]demonstrated that the deficiency ofPCMT1led to progressive epileptic disease or progressive neurodegeneration.[16]Similarly,the role ofPCMT1as a negative regulator in Aβ peptide formations was also documented.Increased β-amyloid production was associated with the knock-down of proteinPCMT1.[17]
DNA methylation is a key factor in regulating synaptic plasticity,therefore affects learning and memory.[18,19]Meanwhile,C3orf14was demonstrated as a methylation regulated gene in methylation arrays research,[20]which was also identified by WGCNA and validated by external data in the current study.However,there is no published study pertaining to its potential role in AD development.
On the other hand,the black module portrayed prominent application in citric acid (TCA) cycle(R-HSA-1428517),respiratory electron transport,mitochondria functions,and metabolism.Deficiencies of mitochondria functions and TCA cycle-related metabolites in AD models were highlighted in various research activities[21,22]and in mitochondria-targeted therapy.[23–25]The administration of TCA cycle-related metabolites could alleviate AD cognitive deficiency.[26,27]Therefore,the results in our study suggested that the black module hub genes may be involved in the TCA cycle and act as potential targets for AD therapy.
ATPase H+transporting V1 subunit A (ATP6V1A),a multisubunit enzyme that mediates acidification of eukaryotic intracellular organelles,is also associated with synaptic vesicle proton gradient generation in brain,energy metabolism,and ATP synthesis.ATP-6V1Awas screened out by co-expression network construction of black module with high network connectivity.Meanwhile,the black module was significantly related to the TCA cycle and mitochondria functions.The external dataset also validated this finding.Therefore,we inferred thatATP6V1Amay play an important role in AD.Research by Fassio found that mutations inATP6V1Aattributed to the onset of developmental encephalopathy with epilepsy,which suggested its role in regulation of neuronal development.[28]However,there had been no study uncovering the potential function of this gene in AD.
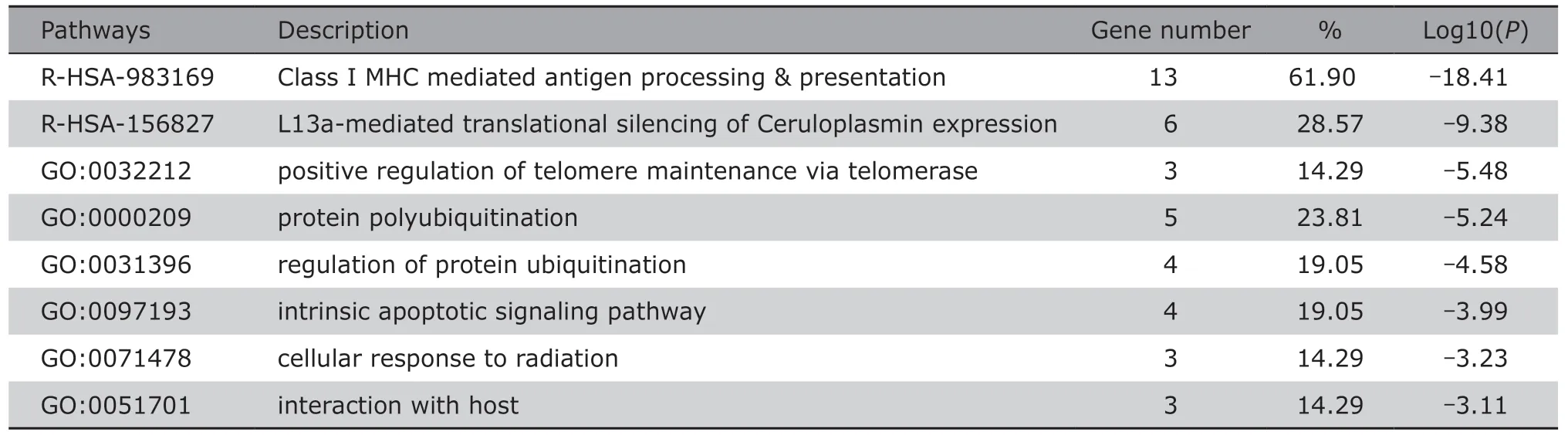
Table 2.GO and KEGG analyses of the sub-network cluster in genes PPI network of black module§
SLC25A14is a mitochondrial uncoupling protein with a great abundance in brain.[29]Meanwhile,Anithaet al.[30]found that the reduction inSLC25A14expression was associated with mitochondrial dysfunction in the autism spectrum disorders,presuming its role in the regulation of mitochondria functions in brain.A homodimeric mitochondrial matrix enzyme encoded by the geneOXCT1plays a central role in extrahepatic ketone body catabolism.[31]It is also obtained as a hub gene in the black module.Above all,we uncovered three hub genes (ATP6V1A,SLC25A14,OXCT1) that may be involved in AD occurrence through pathway of TCA cycle or regulating mitochondria functions.To our best knowledge,no study had reported the potential function of these genes in AD.
In summary,we performed WGCNA on microarray data and found six modules that were highly correlated with AD occurrence.Finally,we identified three hub genes (ATP6V1A,SLC25A14,OXCT1) that may be involved in AD pathogenesis through regulating TCA cycle and may serve as therapeutic targets for AD.
Conflict of interests
The authors declared no conflicting interests.
Supplementary matierials
Figure S1 and Figure S2.
Available online at http://cmsj.cams.cn/EN/10.24920/003695.
 Chinese Medical Sciences Journal2020年4期
Chinese Medical Sciences Journal2020年4期
- Chinese Medical Sciences Journal的其它文章
- Stiff-Person Syndrome Associated with Anti-Glutamic Acid Decarboxylase Autoimmune Encephalitis in a Young Woman:A Case Report
- Ultrasound-Guided Transmuscular Quadratus Lumbar Block Reduces Opioid Consumption after Laparoscopic Partial Nephrectomy
- Detection of Asymptomatic Carotid Artery Stenosis in High-Risk Individuals of Stroke Using a Machine-Learning Algorithm
- CT Texture Analysis:A Potential Biomarker for Evaluating KRAS Mutational Status in Colorectal Cancer
- Bacterial Spectrum and Antimicrobial Profile of Pediatric Blood Stream Infection at a Tertiary Care Hospital in Pakistan
- Study on Image Acquisition of Transthoracic Echocardiography in Mechanically Ventilated ICU Patients