
1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔的合成与表征
2021-01-06 08:17:36刘念陈翔杨柳柳利
湖北大学学报(自然科学版) 2021年1期
刘念,陈翔,杨柳,柳利
(有机化工新材料湖北省协同创新中心(湖北大学),有机功能分子合成与应用教育部重点实验室(湖北大学), 湖北 武汉 430062)
0 引言
由于炔基的直线型结构、高稳定性和π-电子的共轭效应,炔基金属配合物常表现出优异的结构特性,如非线性光学效应[1-2]、荧光[3-6]、光导电性[7-9]、电子传输(分子导线)[10]和液晶性质[11].C≡C键与金属成键最常见的是以σ-键相连,形成线型结构;此外,C≡C键与多个金属通过π键桥联形成结构更加稳定的簇合物[12].
近年来,日本化学家Osawa等[13]报道了一种高效绿光三配位卤化铜(I)配合物,用其组装的OLED的外量子效率(EQE)最大值达到21.3%,可与传统贵金属铱的配合物的外量子效率相媲美,使成本低、丰度高的铜(I)配合物成为替代贵金属配合物的重要选择之一. 为了探索乙炔基与金属的配位作用及配合物的发光性质,我们在前期工作[14-17]的基础上,设计将乙炔基引入有机双膦配体中,具体步骤如图1所示.以4-溴-1,2-二甲氧基苯为原料,通过碘代、Sonogashira交叉偶联反应、脱TMS得到端炔化合物,然后与碘代物再进行一次交叉偶联反应得到1,2-二(2-溴-4,5-二甲氧基苯基)乙炔,最后依次与正丁基锂、二苯基氯化膦反应生成目标产物1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔.用核磁共振谱(1H NMR、13C NMR、31P NMR)、傅里叶变换红外光谱(FT-IR)、高分辨电喷雾质谱(HRMS-ESI)等对目标化合物进行测定和结构表征,进一步确证了目标物的结构.
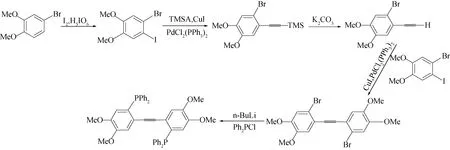
图1 1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔的合成路线
1 实验部分
1.1 仪器与试剂仪器:美国瓦里安AS600(400, 500 MHz)高分辨率核磁共振仪、美国Theromo Scientific Exactive Plus高分辨电喷雾电离质谱仪、BXFI-IR型傅里叶转换红外光谱仪(美国Perkin-Elmet 公司,KBr压片)、101-2AB电热鼓风干燥箱、RE-52AA旋转蒸发仪(上海亚荣生化仪器厂)、SHZ-Ⅲ型循环水式真空泵、恒温加热磁力搅拌器、BSA224S型电子天平(北京赛多利斯仪器系统有限公司)、DZF-602型真空干燥箱、磁力搅拌器和氮气保护装置.
试剂:所有试剂均为市售,试剂纯度均为AR,THF经钠丝预干燥,加热回流至指示剂二苯甲酮呈蓝色后蒸出备用.
1.2 1-溴-2-碘-4,5-二甲氧基苯的合成[18]将4-溴-1,2-二甲氧基苯(10 g, 46.07 mmol)、碘(4.68 g, 18.44 mmol)和高碘酸(2.10 g, 9.21 mmol)溶于50 mL甲醇中,将混合液加热回流9 h,然后冷却至室温,将反应后的混合液倒入硫代硫酸钠溶液(3 g/150 mL)中,有沉淀生成,收集沉淀,采用柱层析(V二氯甲烷∶V石油醚=1∶6)分离纯化,得白色固体1-溴-2-碘-4,5-二甲氧基苯14.5 g,产率92%.1H NMR (400 MHz, CDCl3),δ=7.23 (s, 1H), 7.08 (s, 1H), 3.86 (s, 3H), 3.85 (s, 3H).
1.3 2-(2-溴-4,5-二甲氧基苯基)乙炔基三甲基硅烷的合成[18]在混有1-溴-2-碘-4,5-二甲氧基苯(10 g,29.16 mmol)、二(三苯基膦)二氯化钯(60 mg, 0.085 mmol)、CuI(37 mg, 0.194 mmol)、三乙胺(20 mL)和干燥的四氢呋喃(50 mL)的混合溶液中,滴加三甲基硅乙炔(3.16 g, 32.17 mmol)的干燥四氢呋喃溶液(50 mL),滴加完毕,在室温下反应10 min,再加热回流反应15 h. 反应完全后,冷至室温,用砂芯漏斗抽滤,将滤液收集减压浓缩后得粗品,采用柱层析(V二氯甲烷∶V石油醚=1∶2)分离得白色固体 8.8 g,产率96%.1H NMR (400 MHz, CDCl3),δ=7.01 (s, 1H), 6.96 (s, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 0.27 (s, 9H).
1.4 1-溴-2-乙炔基-4,5-二甲氧基苯的合成[18]将2-(2-溴-4,5-二甲氧基苯基)乙炔基三甲基硅烷(5 g, 15.96 mmol)加入到甲醇和二氯甲烷的混合溶液(60 mL,V∶V=2∶1)中,再加入K2CO3(6.64 g, 48.04 mmol),在室温反应3 h,然后将反应混合物倒入水中,用二氯甲烷萃取3次,经无水硫酸钠干燥后,减压浓缩后得粗品,采用柱层析(V二氯甲烷∶V石油醚=1∶1)分离得到白色固体 3.0 g,产率78%.1H NMR (400 MHz, CDCl3), 7.23 (s, 1H), 7.08 (s, 1H), 3.86 (s, J 3H), 3.85 (s, 3H), 3.29 (s, 1H).
1.5 1,2-二(2-溴-4,5-二甲氧基苯基)乙炔的合成[18]将混有二(三苯基膦)氯化钯(30 mg,0.043 mmol)、CuI(20 mg,0.11 mmol)、1-溴-2-碘-4,5-二甲氧基苯(6.3 g,18.37 mmol)、三乙胺(20 mL)和四氢呋喃(50 mL)的溶液中,滴加1-溴-2-乙炔基-4,5-二甲氧基苯(4 g,16.59 mmol)的四氢呋喃溶液(50 mL),在室温下反应5 min,再加热回流15 h, 反应完毕后冷却至室温,将反应混合物用砂芯漏斗抽滤,滤液减压浓缩后再溶于二氯甲烷,依次用饱和氯化铵溶液、饱和食盐水和水洗涤,分液后收集有机层,无水硫酸钠干燥、过滤、减压浓缩得粗品,柱层析(V二氯甲烷∶V石油醚=1∶4)分离得到黄色固体6.1 g,产率81%.1H NMR (400 MHz, CDCl3),δ=7.07 (s, 2H), 7.06 (s, 2H), 3.90 (s, 12H).
1.6 1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔的合成在氮气保护下将 1,2-二(2-溴-4,5-二甲氧基苯基)乙炔(4.0 g,8.77 mmol)溶于 40 mL无水四氢呋喃中,冷却至-78 ℃,缓慢滴加正丁基锂(2.5 mol/L己烷)溶液(7.72 mL, 19.3 mmol),得到黄色溶液,-78 ℃保温1 h后加入氯化二苯基膦 3.42 mL(18.46 mmol)的THF溶液,继续 -78 ℃保温 5 h,反应溶液呈淡黄色透明,恢复室温后,再用5 mL脱气甲醇猝灭,旋蒸除去溶剂THF,加水用二氯甲烷萃取3次,合并萃取液,用无水硫酸镁干燥,过滤、旋干,通过柱层析色谱分离(V石油醚∶V二氯甲烷=2∶1)得到白色固体产物 1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔3.0 g,产率 51%.1H NMR(400 MHz,CDCl3),δ=7.36~7.28 (m, 20H),6.59 (d,JH-P=4 Hz, 2H), 6.20 (d,JH-P=4 Hz, 2H), 3.79 (s, 6H), 3.50 (s, 6H).13C NMR(100 MHz, CDCl3),δ=148.96, 148.88, 137.26, 137.13, 133.89, 133.69, 131.56, 131.45, 128.56, 128.41, 128.37, 128.34, 121.08, 120.78, 115.06, 114.82, 114.76, 93.76, 55.99, 55.39. 31P NMR (160 MHz, CDCl3),δ=-8.79. HRMS (m/z): calcd for [M+H], 666.208 9; found: 666.207 4.
2 结果与讨论
2.1 FT-IR图谱图2是目标化合物1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔固体粉末态的红外光谱,高频区3 046和3 013 cm-1为芳环C—H键伸缩振动产生的吸收峰,2 958和2 859 cm-1为甲氧基的C—H键的伸缩振动峰,1 596、1 467和1 395 cm-1吸收带为芳环骨架伸缩振动产生的吸收峰,1 270、1 230、1 062 cm-1为C—O键伸缩振动产生的吸收峰.低频区中742、702 cm-1吸收带为一取代芳环C—H键弯曲振动产生的吸收峰. 由于目标化合物结构对称,C≡C的偶极矩为零,在2 100 cm-1左右处未见C≡C特征吸收峰. IR光谱结果与分子结构相符.
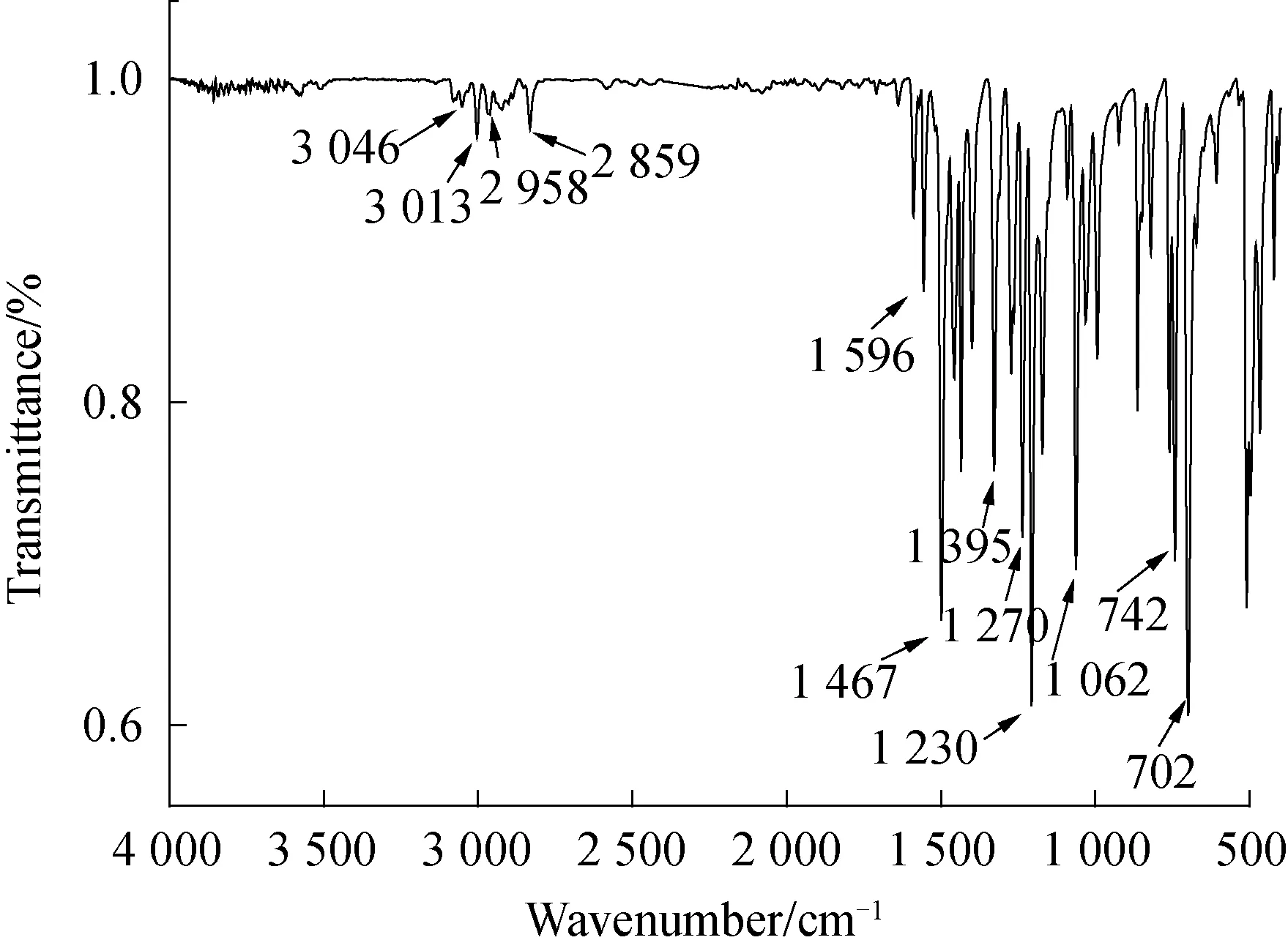
图2 1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔的红外光谱
2.21H NMR图谱图3为目标化合物1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔在CDCl3中的1H NMR图谱,化学位移为7.36~7.28的多重峰对应于两个二苯基膦上4个苯环上的H,四取代苯环上的2个H的化学位移分别为 6.59和6.20, 由于与P原子偶合作用导致裂分为双重峰,四取代苯环上的2个甲氧基化学位移分别为3.79和3.50,每种氢对应的积分面积与结构相符.图中化学位移为1.56处的小峰为水的残留峰.
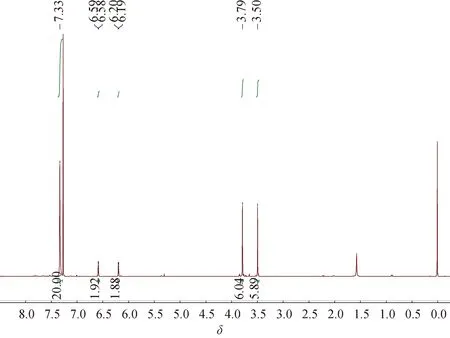
图3 1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔的1H NMR图谱
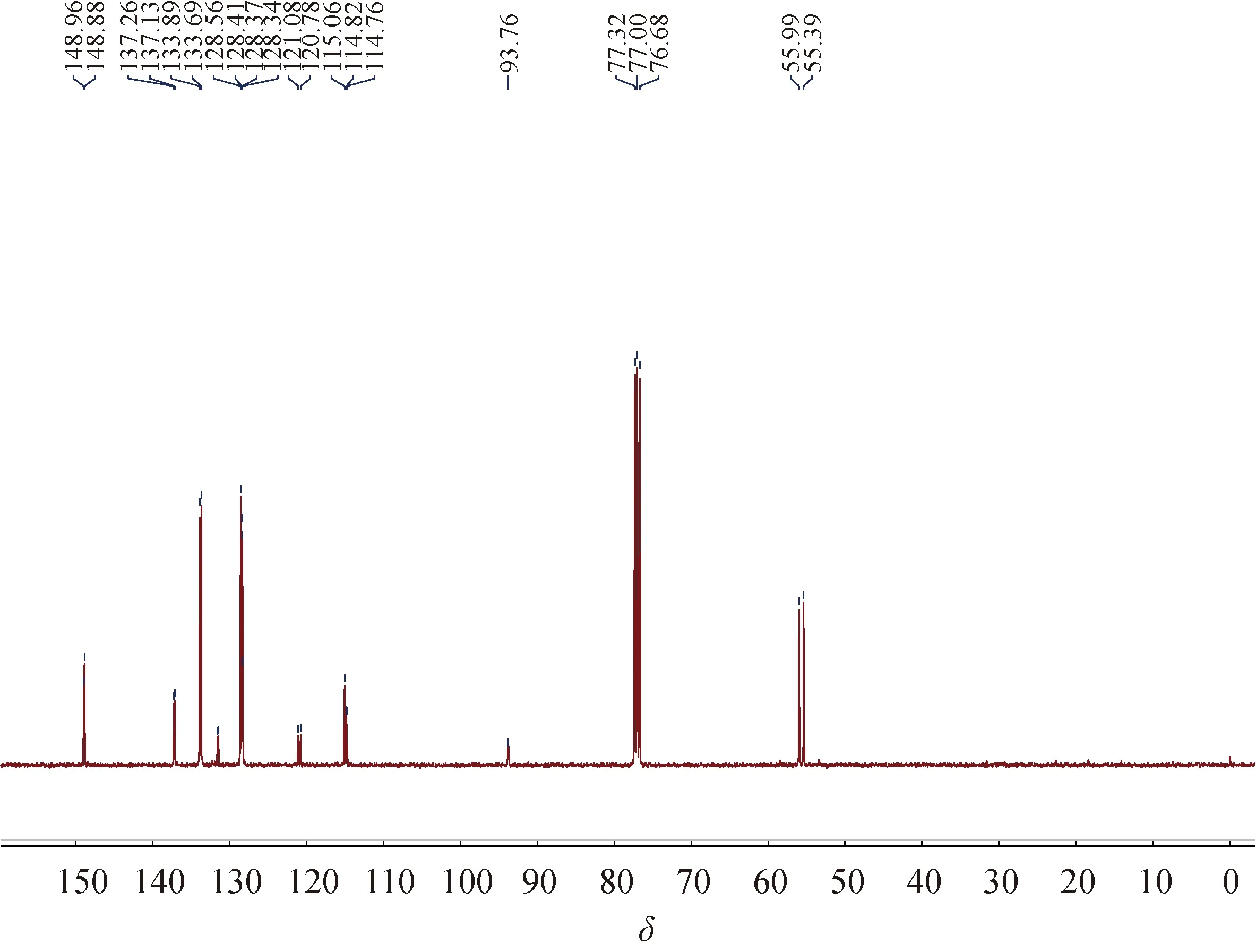
图4 1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔的13C NMR图谱
2.313C NMR图谱图4为1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔在CDCl3中的13C NMR图谱.四取代苯环及二苯基膦上苯环上的碳原子的化学位移在 148.96~114.76 之间出现共振信号峰,分别对应于苯环上的 36个碳原子,表明结构具有对称性. 两个四取代苯环上4个甲氧基上的碳的核磁共振信号峰有2个,化学位移分别为 55.99和55.39,两个三键碳的核磁共振信号峰只有1个,其化学位移为 93.76, 同样也证明了化合物结构的对称性.
2.431P NMR图谱图5为1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔在CDCl3中的31P NMR图谱.在化学位移为-8.79 处有一个共振信号峰,为1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔中C—P共振信号峰,证明了化合物的结构具有对称性.
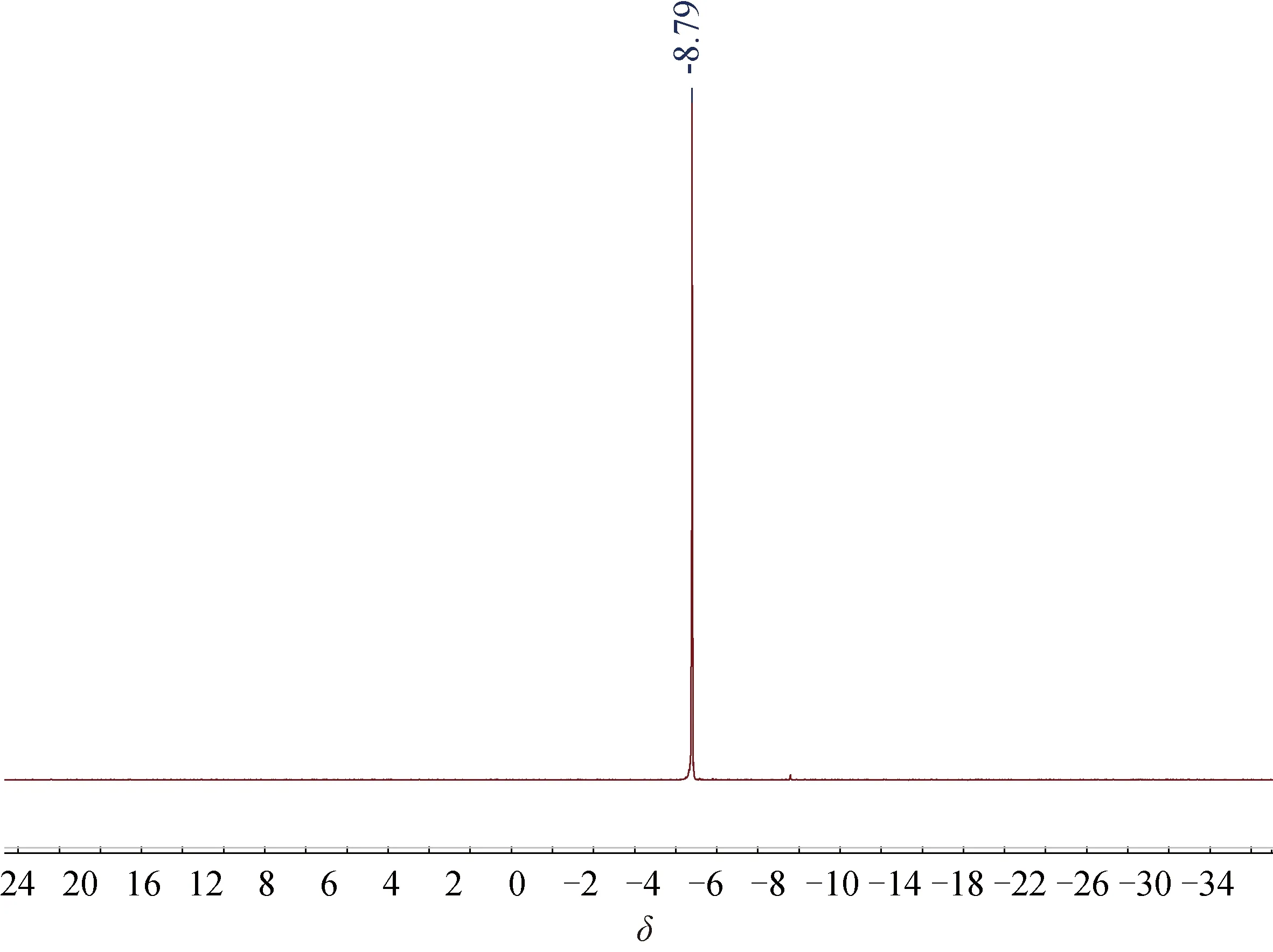
图5 1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔的31P NMR图谱
2.5 高分辨质谱图6为目标化合物1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔以电喷雾(ESI)为电离方式的高分辨质谱,在m/z=666.207 4处可见强烈的信号峰,为目标化合物的分子离子峰,与计算值666.208 9相符合,可进一步确定目标化合物的结构.
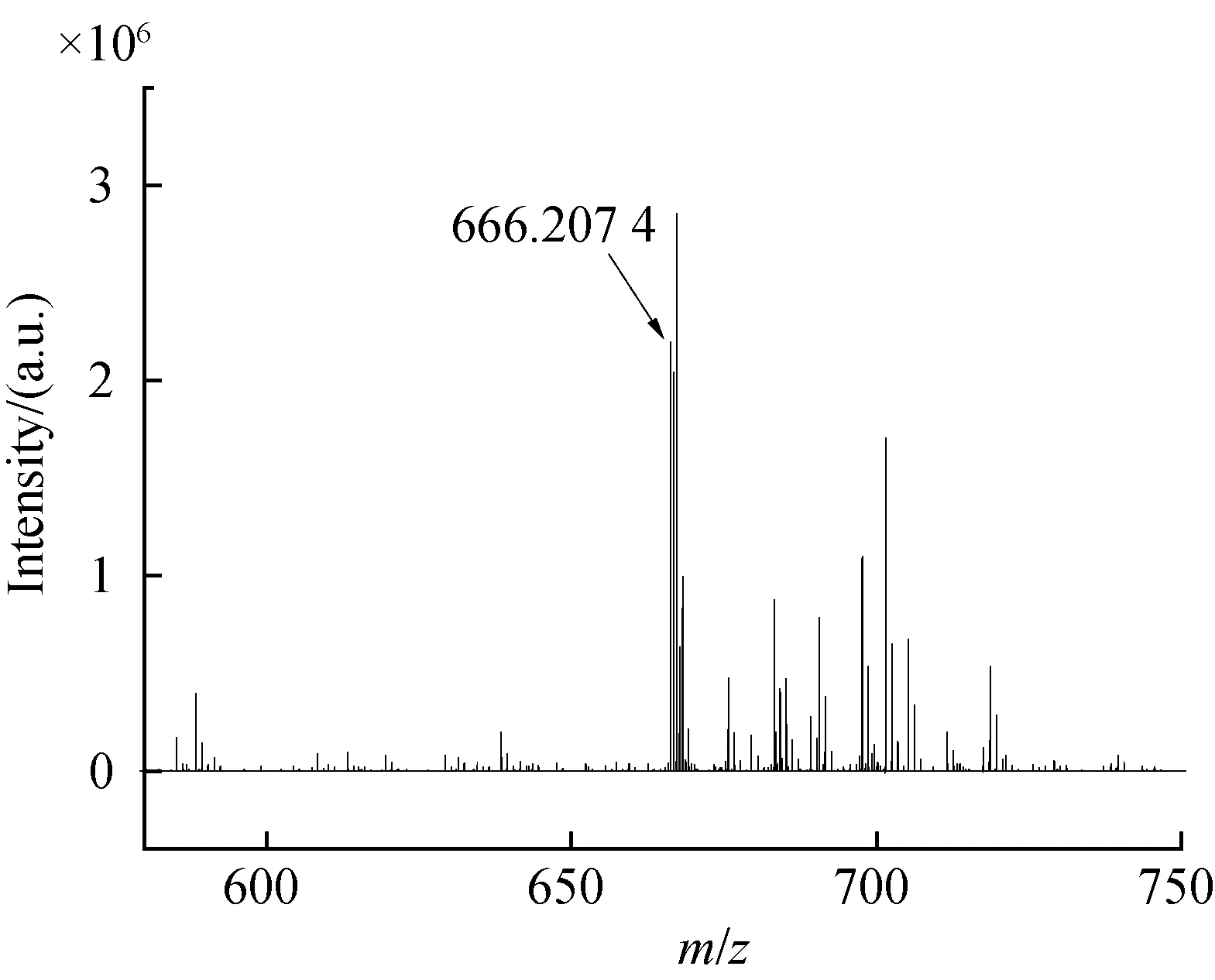
图6 1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔的MS图
2.6 反应机理在目标化合物的合成路线中,有两步采用Sonogashira交叉偶联反应.与文献[18]已报道的合成方法相比, 我们使用的催化剂Pd(PPh3)2Cl2和CuI, 其用量百分比分别仅为0.26%和0.66%,比文献中报道的Pd(PPh3)2Cl2和CuI催化剂的用量百分比(4%和8%)分别降低了15倍和12倍,在保证反应具有较高收率的基础上,大幅降低了成本,同时简化了后续的分离过程. 可能的偶联反应机理如图7所示:首先,Pd(PPh3)2Cl2与端炔在三乙胺碱性条件下生成钯炔化合物(PPh3)2Pd(C≡CR)2和三乙胺的盐酸盐,然后经过还原消除生成(PPh3)2Pd0,只有零价的钯才能有效催化这个反应,但零价的钯对氧气比较敏感,所以这个反应需要在惰性气体保护下进行. 有碘化亚铜作共催化剂的Sonogashira反应发生了双环反应(图7),主要包含三步反应:1)芳基碘代物氧化加成到Pd0化合物上;2)钯催化的环和亚铜做共催化剂的环相结合,速率控制步骤在于铜环下产生的铜炔与钯催化环的中间体RPd(PPh3)2I发生金属转移反应得到RPd(PPh3)2(C≡CR);3)通过顺反异构(图中省略)和还原消除生成产物. 显然,该反应体系中,CuI是助催化剂,三乙胺作为碱性溶剂与反应试剂,有助于将端炔的H移去.

图7 Sonogashira交叉偶联反应的可能机理图示
3 结论与展望
以4-溴-1,2-二甲氧基苯为原料,通过碘代、Sonogashira交叉偶联反应、脱TMS得到端炔化合物,然后与碘代物再进行一次交叉偶联反应得到1,2-二(2-溴-4,5-二甲氧基苯基)乙炔,最后依次与正丁基锂、二苯基氯化膦反应生成目标产物1,2-二[(2-二苯基膦基-4,5-二甲氧基)苯基]乙炔,用核磁共振谱、傅里叶变换红外光谱、高分辨电喷雾质谱等对目标化合物进行了测定和结构表征,确证了目标物的结构.采用的合成路线具有操作简单、反应条件温和、产率高等优点.尤其是在Sonogashira交叉偶联反应中使用了比已报道的文献值低15倍和12倍的金属钯及铜催化剂,大幅降低了成本.另外,合成的目标化合物为炔基双膦配体,具有良好的配位能力,预计将获得结构新颖、具有良好发光性质的金属配合物, 并应用于光电器件等研究领域.
猜你喜欢
山东化工(2019年7期)2019-04-27 07:39:28
心肺血管病杂志(2019年1期)2019-04-22 01:11:58
科普创作(2018年1期)2018-11-30 05:11:40
中国资源综合利用(2017年1期)2018-01-22 02:44:25
中国塑料(2017年2期)2017-05-17 06:13:27
当代化工研究(2016年9期)2016-03-20 16:22:19
电源技术(2016年9期)2016-02-27 09:05:25
中国塑料(2015年8期)2015-10-14 01:10:49
苏州科技大学学报(工程技术版)(2015年3期)2015-02-28 16:21:12
自动化博览(2014年8期)2014-02-28 22:33:06
