羟基封端聚丁二烯的临界点色谱定量方法研究
2020-12-25 02:41汪明芳贾强强王月荣章弘扬
色谱 2020年2期
汪明芳,贾强强,王月荣,章弘扬,张 敏,胡 坪*
(1.上海市功能性材料化学重点实验室,华东理工大学化学与分子工程学院,上海 200237;2.上海市新药设计重点实验室,华东理工大学药学院,上海 200237)
羟基封端的聚丁二烯(HTPB)是一种新型的重要工业原料,在医疗器械、电子工业、能源、化工等行业有着广泛应用[1-4]。HTPB常被用于聚氨酯基体的胶黏剂和涂料中,与矿物油、蓖麻油等复配形成二组分胶黏剂或涂料的组分之一,再与另一组分异氰酸酯反应,用于电子元器件等的黏结与封装[5,6]。因此,HTPB成分的测定对于产品应用过程的失效分析、未知产品的成分剖析以及产品的生产过程及质量控制都有重要意义。然而在混合体系中,聚合物的定量分析一直是一个具有挑战性的难题,特别是当宽分布的HTPB与共存组分的相对分子质量相近、结构相似时,HTPB组分的分离与定量检测更加困难。
目前混合体系中聚合物的定量分析方法有元素分析法、红外光谱法、紫外光谱法[7-11]等,然而这些分析方法需要聚合物含有特征元素或官能团。近年也有将MALDI-TOF用于聚合物测定的报道[12-17],然而该技术用于定量分析时重复性较差,存在组成与相对分子质量的歧视,且需要使用同位素内标,因此应用并不广泛。目前通用的聚合物定量分析方法是核磁共振法[18,19],但其中的氢谱因化学位移范围较窄,谱线易发生重叠,导致定量不准确,因此需要与分离技术结合进行定量分析[20-22];碳谱具有较高的分辨率,但13C的自然丰度较小,测定灵敏度较低,定量13C测量时间长,运用较少。另外,基于质谱和核磁的聚合物定量方法还存在仪器价格贵、普及率不高的缺点。
临界点色谱法是近年来提出的一种介于体积排阻色谱和吸附分离色谱之间的过渡态分离方法。依据色谱热力学和动力学原理,聚合物在色谱洗脱过程中受热焓控制时,宽分布聚合物中的大分子与固定相间的相互作用大,保留时间长,而小分子与固定相作用弱,保留时间短;而当无热焓的作用、聚合物洗脱过程完全受热熵主导时,聚合物按相对分子质量大小被洗脱。以上两种情况下,聚合物的色谱峰都会产生严重的展宽,这也是聚合物一般只能用排阻色谱分离,并仅用于其相对分子质量测定的主要原因。当聚合物在临界点或近临界点色谱条件时,因洗脱过程中热焓和热熵的作用相互补偿,几乎没有吉布斯自由能的变化,这时色谱“无视”聚合物相对分子质量的大小,不同相对分子质量的同种聚合物几乎在同一保留时间被洗脱,聚合物色谱峰宽将很窄[23-26]。因此,在特定的流动相组成和温度下,在特定的色谱固定相上,聚合物会呈现与相对分子质量无关的保留行为。利用这种特定的临界点色谱条件,即可实现聚合物混合物以及均聚物与共聚物的分离[27,28]。目前,临界点色谱已被应用于基于成分分布、立体结构分布等特征的聚合物分离分析中[29-32],但尚未见用该方法对混合物中的聚合物进行准确定量的报道。
本文研究了HTPB聚合物的临界点色谱条件,并对HTPB与矿物油复配形成的胶黏剂进行了分离和定量分析。临界点色谱方法对聚合物的定量分析具有误差小、灵敏度高、对仪器设备要求不高的特点,在混合体系中聚合物的定量分析中有应用前景。
1 实验部分
1.1 仪器、试剂与材料
Waters ACQUITYR ArcTM高效液相色谱仪配Waters 2424 ELS,Waters 2695凝胶色谱仪配Waters 2414折光检测器(美国Waters公司);Milli-Q超纯水纯化系统(美国Millipore公司);核磁共振波谱仪(德国布鲁克光谱仪器公司);分析天平(瑞士梅特勒-托利多国际贸易(上海)有限公司)。
乙腈(ACN,色谱纯,德国默克化工技术上海有限公司);四氢呋喃(THF,色谱纯)、氘代氯仿(纯度99.9%)(美国西格玛奥德里奇上海贸易有限公司);宽相对分子质量分布的HTPB溶液(工业级,相对分子质量为8191 Da;聚合物分散性指数(PDI)为2.1,法国阿科玛公司);矿物油(工业级,上海麦克林生化科技有限公司);市售双组分聚氨酯胶黏剂(深圳爱乐特投资有限公司)。窄相对分子质量分布的HTPB溶液(峰尖相对分子质量(Mp)为13 330、7 828、2 595、1362 Da)由本实验室用凝胶色谱法自制,相对分子质量用标准的聚苯乙烯校正获得。0.2 μm聚四氟乙烯(PTFE)膜购自美国赛默飞世尔科技(中国)有限公司。
1.2 HTPB临界点色谱条件的探索
1.2.1样品溶液的配制
以四氢呋喃做溶剂,分别配制质量浓度为1.5 g/L的4个窄相对分子质量分布的HTPB溶液,用于液相色谱临界点条件的探索。
1.2.2四氢呋喃-乙腈流动相体系的临界点色谱条件
色谱柱:Diamonsil®C18柱(200 mm×4.6 mm,5 μm,迪马科技);流速:1 mL/min;进样量:2 μL;柱温:35 ℃;检测器:蒸发光散射(ELSD)检测器;漂移管温度:80 ℃;雾化器温度:60 ℃;气流压力:206.8 kPa。在流动相四氢呋喃和乙腈体积比分别为60∶40、70.7∶29.3和80∶20时,测定窄相对分子质量分布HTPB的色谱保留行为。
1.2.3四氢呋喃-水流动相体系的临界点色谱条件
在流动相四氢呋喃和水体积比分别为86∶14,92∶8和96∶4时,测定窄相对分子质量分布的HTPB溶液色谱保留行为。其余条件与1.2.2节相同。
1.3 HTPB的临界点色谱定量分析
1.3.1标准溶液的配制
称取适量宽相对分子质量分布的HTPB,用四氢呋喃配制成质量浓度分别为46.7、91.5、134.7、176.3和216.4 mg/L的标准溶液。
1.3.2样品配制
取宽相对分子质量分布的HTPB和矿物油,按照26∶74的质量比充分混匀。称取混合后的样品,用四氢呋喃配制成含91.5 mg/L HTPB的样品溶液,用于定量方法的回收率测定。
取适量的市售胶黏剂未知样品,用四氢呋喃配制成292.4 mg/L的样品溶液,用于实际样品中HTPB的定量分析。
1.3.3色谱条件
流动相为四氢呋喃-乙腈(70.7∶29.3,v/v),其余色谱条件同1.2.2节。
1.3.4核磁定量分析
取1.3.2节中的HTPB和矿物油混合样品以及市售胶黏剂未知样品适量,分别配制成质量浓度为20.0 g/L的氘代氯仿溶液,用于核磁共振氢谱分析。
2 结果与讨论
2.1 HTPB的临界点色谱流动相条件筛选
流动相的组成是实现临界点色谱分离的关键,一般临界点色谱使用混合流动相来达到临界点条件,即利用聚合物的良溶剂和不良溶剂的混合来调整聚合物的保留行为。其中一种溶剂利于聚合物吸附于固定相上,另外一种溶剂则用于聚合物的脱附。例如,HTPB在四氢呋喃中有很好的溶解能力,能使HTPB从色谱固定相上按体积排阻方式洗脱,当不良溶剂乙腈或者水加入流动相中后,就大大降低了HTPB分子从固定相上洗脱过程的熵的变化。在临界点条件下,热焓与热熵的变化相互补偿抵消,不同相对分子质量的HTPB在同一时间被洗脱,色谱峰窄。本文选用HTPB聚合物的不良溶剂乙腈和水分别与四氢呋喃混合,作为聚合物临界点色谱的流动相,研究了两种流动相体系下HTPB的保留规律及临界点色谱条件。
4个窄相对分子质量分布的HTPB在用不同体积比的四氢呋喃-乙腈为流动相时的保留行为如图1a所示。当四氢呋喃-乙腈体积比为80∶20时,大分子的HTPB先被洗脱,小分子的HTPB后被洗脱,表明在该条件下,HTPB的分离主要基于体积排阻机理。当四氢呋喃-乙腈的比例降至60∶40时,HTPB保留时间随相对分子质量的增大而延长,此时,聚合物与固定相间的吸附作用主导分离的过程。当四氢呋喃-乙腈的比例为70.7∶29.3时,不同相对分子质量的HTPB几乎在同一时间洗脱,该流动相比例即为HTPB色谱分离的临界点条件。
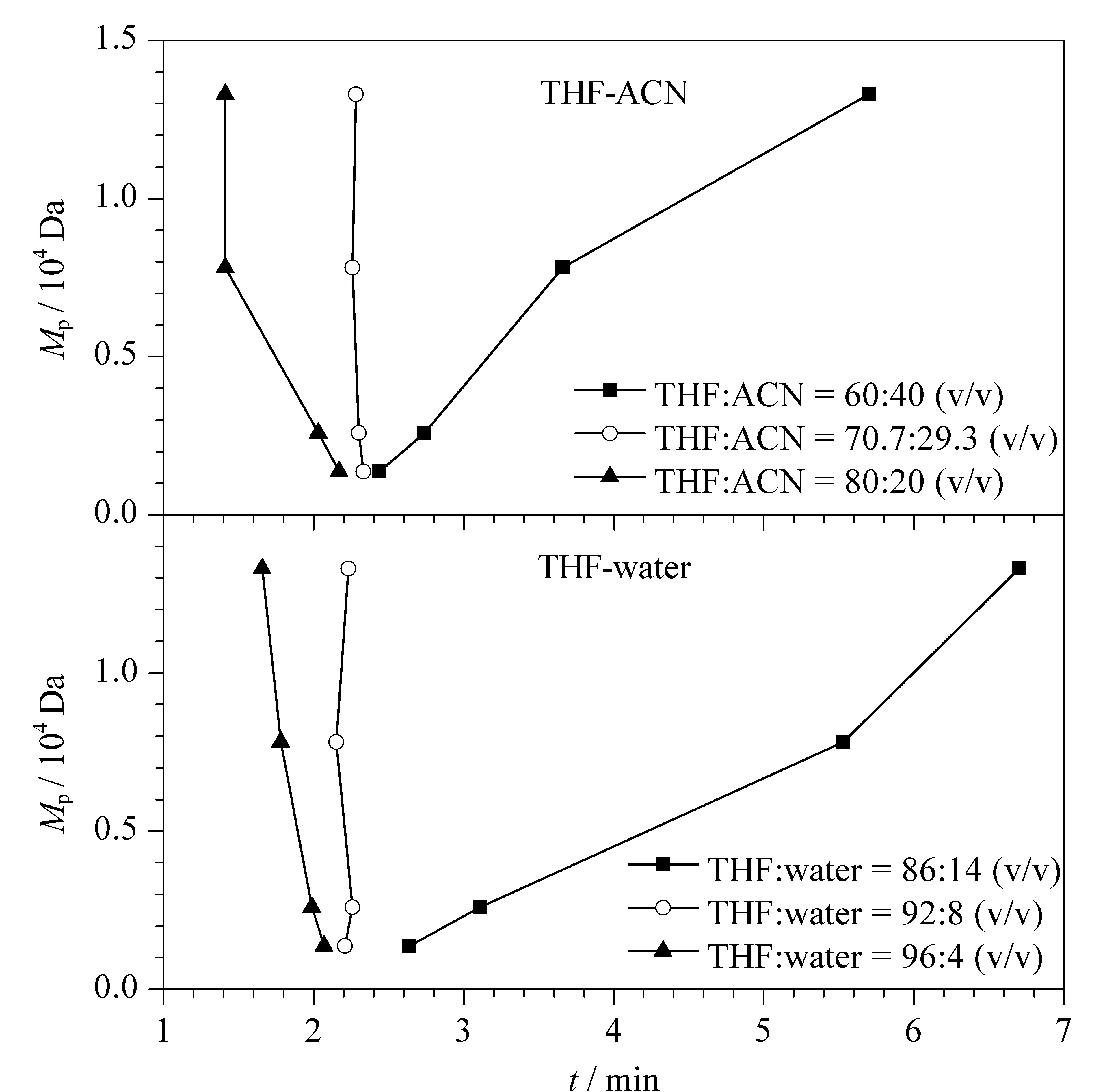
图1 窄相对分子质量分布的HTPB在用不同比例(a)四氢呋喃和乙腈、(b)四氢呋喃和水为流动相时的保留行为Fig.1 Retention behavior of narrowly distributed hydroxyl terminated polybutadiene (HTPB)using tetrahydrofuran (THF)-acetonitrile (ACN)and THF-water in different ratios as the mobile phases Peak relative molecular mass (Mp):13330,7828,2595,and 1362 Da.
当四氢呋喃和水作为流动相时,4个相对分子质量HTPB的保留值与流动相比例的关系如图1b所示。四氢呋喃-水的体积比为92∶8时,不同相对分子质量的HTPB几乎在同一保留时间下洗脱;当四氢呋喃比例减小到86%时,大分子的HTPB保留时间长,呈现以吸附模式主导的分离;而当四氢呋喃的比例增大到96%时,出现明显的体积排阻模式保留规律。以上研究表明,在四氢呋喃-水体系中,体积比为92∶8是HTPB聚合物的临界点色谱条件。
由此可见,四氢呋喃-乙腈(70.7∶29.3,v/v)和四氢呋喃-水(92∶8,v/v)均可作为HTPB分离的临界点色谱条件。然而,以四氢呋喃-水作流动相时,当水比例稍有增大(14%),聚合物保留时间即显著延长。此时,在临界点附近,流动相比例的微小波动可能对聚合物大分子保留值有较大影响,方法的耐受性将变差。因此,选择四氢呋喃-乙腈(70.7∶29.3,v/v)体系作为HTPB的临界点色谱条件。

图2 四氢呋喃溶剂空白和宽分布HTPB的临界点色谱图Fig.2 Chromatograms of THF blank and broadly distributed HTPB under the critical conditions
2.2 宽分布HTPB和矿物油混合组分的临界点色谱分离
工业产品中使用的HTPB多为宽相对分子质量分布,用常规的反相或正相液相色谱分析时,色谱峰展宽严重,导致难以实现分离和定量测定。根据2.1节研究结果,在临界点条件下,多个不同相对分子质量的HTPB在同一保留时间被洗脱,因此,在该条件下宽相对分子质量分布的HTPB不会发生展宽,可实现聚合物混合试样的高选择性分离。以ELSD为检测器,C18为固定相,溶剂空白和宽相对分子质量分布HTPB的临界点色谱图见图2。由图可见,宽相对分子质量分布HTPB的半峰宽仅为0.2 min左右,与反相色谱中小分子化合物的峰宽相近,表明临界点色谱的保留行为与被分离聚合物的相对分子质量无关,体现了该方法“无视”聚合物相对分子质量大小的特性。
图3a和3b分别是宽相对分子质量分布HTPB与矿物油混合组分的体积排阻色谱图和临界点色谱图。由图可见,重均相对分子质量为8191 Da的宽分布HTPB在排阻色谱中呈现为一宽峰,与相对分子质量小于1 000的矿物油并不能达到完全分离。而在临界点色谱图中,基于HTPB和矿物油化学结构和极性的不同,实现了两者的良好分离,从而为混合物中HTPB组分的定量分析提供了有效的方法。

图3 宽相对分子质量分布HTPB与矿物油混合物的色谱分析图Fig.3 Chromatograms of a mixture of broadly distributed HTPB and mineral oil a.size exclusion chromatography (SEC);b.liquid critical condition chromatography (LCCC).
2.3 HTPB组分的线性范围、检出限及精密度
配制系列宽分布HTPB的标准溶液,以质量浓度(X,mg/L)的对数为横坐标,以分析物对应的峰面积(Y)的对数为纵坐标绘制标准曲线,得到线性回归方程lgY=1.26lgX+4.26。结果表明,HTPB在46.7~216.4 mg/L范围内线性关系良好,相关系数(r)为0.997。将HTPB标准溶液不断稀释至其信号分别为噪声的3倍和10倍,测得HTPB的检出限为4.2 mg/L,定量限为9.2 mg/L。
对质量浓度为91.5 mg/L的HTPB溶液连续进样测定6次,计算得到其峰面积的相对标准偏差(RSD)为1.4%,表明仪器精密度良好。
2.4 回收率
以自制的HTPB和矿物油混合组分为样品,加入3个浓度水平的HTPB,进行加标回收试验,每个加标水平平行测定6次,回收率见表1。结果表明,HTPB在自制混合组分中的平均加标回收率为89.2%~101.1%,RSDs为0.49%~0.66%。说明该方法具有良好的准确度和重复性,可用于工业产品中HTPB的日常定量检测。

表1 在HTPB和矿物油混合物中HTPB组分的平均加标回收率和相对标准偏差(n=6)Table 1 Average spiked recoveries and RSDs of HTPB in the blends of HTPB and mineral oil (n=6)
2.5 实际样品分析
应用已建立的临界点色谱方法分别对自配的HTPB和矿物油混合样品以及市售胶黏剂未知样品进行了分析,并将分析结果与核磁共振氢谱(1H nuclear magnetic resonance spectroscopy,1H-NMR)测定结果进行比较(见表2)。结果表明,用临界点色谱法检出自配样品中HTPB含量为26.8%,与实际含量26.0%的相对误差为3.1%,且3次平行测定结果的RSD为1.4%,重复性良好。而利用1H-NMR对自配样品中HTPB的测定结果为38.7%,与实际含量的相对误差高达48.8%。由此可见,临界点色谱对混合样品中HTPB的定量误差远小于核磁共振法,定量结果准确可靠。

表2 LCCC和1H-NMR两种方法对HTPB和矿物油混合物以及未知样品中HTPB含量的测定(n=3)Table 2 Determination of HTPB in the blends of HTPB and mineral oil and an unknown sample by LCCC and 1H nuclear magnetic resonance spectra (1H-NMR)methods(n=3)
-:the value is not available for unknown sample.
采用同样的1H-NMR方法对市售双组分黏结剂中HTPB的含量进行了测定,结果为26.6%,RSD为1.2%(n=3),而核磁共振法的测定结果为41.8%。
3 结论
本文对HTPB聚合物的临界点色谱方法进行了研究,分别考察了四氢呋喃-乙腈和四氢呋喃-水作为流动相体系时HTPB的临界点色谱条件。在临界点色谱条件下,不同相对分子质量的窄分布HTPB在同一保留时间被洗脱。以四氢呋喃-乙腈(70.7∶29.3,v/v)为临界点色谱流动相,成功分离了宽相对分子质量分布的HTPB和矿物油混合物组分,并且对混合物中HTPB进行了定量分析。结果表明,相比于传统的核磁氢谱方法,采用临界点色谱法对混合物中的HTPB进行定量检测具有方法简便、定量准确、重复性好等优点,可用于工业上HTPB产品的质量控制、未知产品剖析和产品失效分析。
猜你喜欢
化工设计(2022年4期)2023-01-02
海洋通报(2022年4期)2022-10-10
贵州科学(2022年4期)2022-09-05
发明与创新(2020年5期)2020-05-06
发明与创新·大科技(2020年2期)2020-04-17
科学导报(2019年73期)2019-12-20
现代盐化工(2019年4期)2019-09-10
造纸化学品(2018年5期)2018-01-31
政工学刊(2017年2期)2017-02-20
中成药(2014年10期)2014-02-28