
SLC16A2基因新发变异致Allan-Herndon-Dudley综合征1例报告并文献复习
2020-12-23 06:54:02贾倩芳周福军崔清洋
临床儿科杂志 2020年12期
贾倩芳 周福军 崔清洋
新乡医学院第一附属医院儿科(河南卫辉 453100)
Allan-Herndon-Dudley综合征 (AHDS,OMIM#300523),是一种X染色体隐性遗传病,1944年首次报道[1]。2004年编码单羧酸甲状腺激素转运蛋白8(MCT 8)的SLC 16 A 2基因变异被确认可导致AHDS[2]。AHDS为罕见的遗传性疾病,可导致神经系统和甲状腺功能的损害,其主要临床表现为男性患儿自婴儿期开始的肌张力低下、进食困难和从轻度到重度的发育迟缓/智力残疾,发展为之后的锥体束征,髓鞘发育延迟,锥体外系表现(肌张力障碍,舞蹈手足徐动症,阵发性运动障碍),运动功能减退,面具面容和具有耐药性的癫痫发作,易咳呛和发生呼吸道感染,但一般在儿童早期不易观察到生长障碍。其他尚有甲状腺功能减退和特征性甲状腺试验,即三碘甲状腺原氨酸(FT3)升高、游离四碘甲状腺原氨酸(FT4)降低及促甲状腺激素(TSH)无异常[3]。大多数杂合子女性无临床症状,但约25%有甲状腺功能异常。AHDS预后不佳,常因呼吸道反复感染而死亡。目前尚无AHDS发病率报道,国际上已报道100余例,国内仅为个案报道。本文回顾分析1例AHDS患儿的临床资料及基因检测结果。
1 临床资料
患儿,男,4月龄。3月龄时发现其头部后仰,清醒时为著,伴奶量下降及偶咳。入院前4天上述症状加重,伴喉鸣,就诊于当地县医院,诊断为“支气管肺炎、营养不良、遗传代谢病可疑”;患儿血培养示施氏假单胞菌生长,予抗感染治疗,治疗效果差而转入新乡医学院第一附属医院。患儿系G1P1,足月剖宫产,出生体质量2.3 kg,出生无异常。父母均体健,非近亲结婚;母亲FT3 4.07 pg/mL,FT4 0.58 ng/dL,超敏TSH 2.25 uIU/mL,提示甲状腺功能减退可能。体格检查:体质量5.6 kg,身长56 cm;双肺呼吸音粗糙,可闻及中等量痰鸣音;心、腹无异常;双下肢肌张力增高,四肢肌力5级,生理反射存在,病理反射未引出;患儿追视、追听尚可,双手可居中线位活动,双下肢屈曲,非对称性紧张性颈反射(asymmetric tonic neck reflex,ATNR)姿势存在,可瞬间抬头,踏步反射仍存在,拇指内收,头部后仰,尖足,围巾征不过中线,跟耳征阳性,可逗笑。实验室检查:血常规白细胞(WBC)6.8×109/L,红细胞(RBC)3.36×1012/L,血红蛋白(Hb)93g/L,血小板(PLT)295×109/L;超敏C反应蛋白(CRP)、血氨、乳酸、肝肾功能、心肌酶、电解质均未见异常;FT3 6.63 pg/mL,FT4 0.69 ng/dL,超敏TSH 2.073 uIU/mL;血串联质谱及尿气相色谱-质谱均未见异常。胸部CT示两肺背侧胸膜下区可见带状淡薄密度增高影,边缘欠清,两肺坠积性肺炎改变可能。24小时视频脑电图未见异常;头颅MRI示双侧额顶部脑外间隙略增宽;内囊前肢无明显髓鞘化;双侧额叶部分脑回稍宽大。心脏及双侧髋关节彩超均未见异常。入院后诊断:发育迟缓原因待查,支气管肺炎,蛋白质营养不良。
患儿入院后予抗感染、雾化、康复等综合治疗后,头部后仰减轻,肺部痰鸣音减少。复查胸部CT无明显改变,复查血常规及超敏CRP未见异常,但住院治疗 1个月后肺部痰鸣音仍未消退。予纤维支气管镜检查示支气管内膜炎症;肺泡灌洗液培养金黄色葡萄球菌及耐甲氧西林葡萄球菌均阳性。复查甲状腺功能FT3 9.04 pg/mL,FT4 0.522 ng/dL,超敏TSH 4.54 uIU/ml。升级抗生素为利奈唑胺加强抗感染,补充左旋甲状腺素片。治疗3天后,患儿肺部痰鸣音减轻。住院期间患儿的呼吸道感染再次加重,家属要求自动出院。
因患儿反复呼吸道,抗感染治疗效果欠佳,且生长发育落后,不能排除肺囊性纤维化及纤毛不动综合征。经医学伦理审核以及家属知情同意后,采集患儿和父母的外周血进行全外显子基因检测。使用软件 CASAVA(1.8.2)将原始的数据转化为可识别的碱基序列,获得靶向区域变异位点的信息。通过在线生物预测软件分析蛋白质损伤,获取变异位点。SLC16A2基因的第一外显子引物:正向的引物序列 AGGCAGACCAGGAACAGCAG;反向的引物序列GCCACTCACCTGCTTGGAAC。结果发现,患儿SLC 16 A 2基因第1外显子存在c.193 delC半合子变异,该变异可导致第65号氨基酸由脯氨酸变为精氨酸并发生移码,在移码后的第19个氨基酸处终止(p.Pro65ArgfsTer19),可能造成蛋白质功能遭受严重影响,该变异位点的致病性尚未见文献报道(参考数据库 HGMD Pro及 PubMed)。该变异不归类为多态性变化,在人群中发生的频率极为低下(参考数据库1000Genomes和dbSNP)。家系验证结果表明,父亲该基因位点为野生型,母亲为该基因位点杂合携带,符合X染色体隐性遗传的规律,见图1。根据美国的医学遗传学与基因组学会(ACMG)联合美国的分子病理学会(AMP)2015年制订的基因序列变异解释的标准和指南进行致病性分析[7]。SLC16A2基因 c.193delC的致病性:①当某基因致病机制为功能缺失(Loss-offunction,LOF)时,该基因上的无义变异、移码变异、±1或 2位置的剪切变异、起始密码子变异、单个或多个外显子缺失(非常强的致病性的证据,PVS1);②c.193delC变异通过比对千人的基因组数据库(1000 Genomes)、人类基因的突变数据库(HGMD)未见它的收录(中等的致病性的证据,PM2)。综合上述c.193delC变异的证据的强度为 “PVS1+PM2”,判断为造成患儿发病的疑似致病性变异。
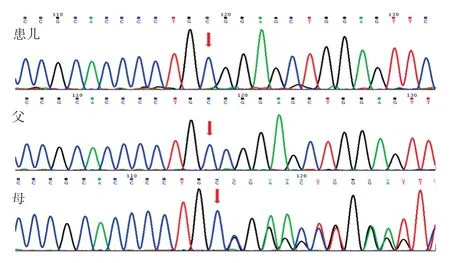
图1 SLC16A2 基因c.193delC变异测序图
全外显子测序检测到患儿DNAH 1基因母源性的c.3853 C>T和c.1321 G>C杂合变异及父源性的c.1138 G>A杂合变异,父母均为杂合状态,符合常染色体隐性遗传方式,而原发性纤毛运动障碍患者常表现为反复呼吸道感染、慢性支气管炎、慢性鼻窦炎、慢性中耳炎及支气管扩张等,患儿已有2次呼吸道感染病史,胸部CT未发现支气管扩张,且ACMG联合 AMP综合判断DNAH1基因c.3853C>T、c.1321 G>C和c.1138 G>A位点的致病性为临床意义不明,从目前数据尚不能认为DNAH 1基因c.3853C>T、c.1321G>C和c.1138G>A变异位点具有致病性。正常序列的SLC16A2蛋白的拓扑结构图(图2),患儿在箭头位置发生移码变异,导致蛋白丢失主要的跨膜结构,从而影响蛋白功能。
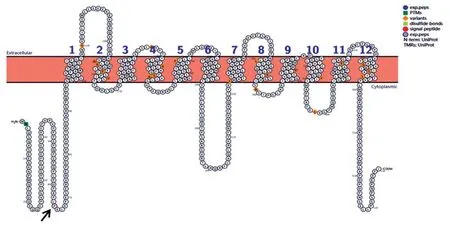
图2 正常序列的SLC16A2蛋白的拓扑结构图
结合患儿临床表现、甲状腺功能及基因检测分析,确诊为SLC16A2基因c.193delC变异所致AHDS。患儿予口服甲状腺素片至今,复查甲状腺功能,FT3 9.72 pg/mL,FT4 0.79 ng/dL,超敏TSH 0.313 uIU/mL;目前在继续治疗中。
2 讨论
SLC 16 A 2基因定位于Xq 13.2,基因组全长约112.46 kb,包含6个外显子和5个内含子,外显子长度约4161 bp,编码539个氨基酸的MCT8蛋白。MCT8蛋白是由12个跨膜结构域组成,是介导甲状腺激素T3进入胞内的特异性转运体,在肝、肾、垂体、甲状腺和脑组织广泛表达。目前业已报道的SLC16A2基因变异大约80种,包括39种错义变异或无义变异,3种剪接变异,10种小片段缺失变异,12种小片段插入,14种大片段缺失,2种大片段插入[4]。
目前认为,SLC16A2基因变异导致的MCT8蛋白部分缺乏或完全缺失,可能造成MCT8蛋白和细胞膜及甲状腺激素的结合障碍,导致甲状腺激素在大脑里的神经元、少突细胞分布下降或完全缺失,大脑神经元及少突细胞的转录和翻译受到影响,导致神经细胞和少突细胞的形成、迁移及分化的成熟障碍,从而出现一系列神经系统症状。此外,MCT8蛋白的缺陷尚可导致甲状腺组织脱碘酶D1及D2的表达升高,T3由T4转换的增加,导致外周血的T4下降而T3的增高,后者致外周的组织表现为代谢增高的状态,糖异生和糖酵解增强,脂肪合成及降解增强,外周组织和骨骼肌蛋白质分解增快,患儿可有如瘦弱及脸型变长等[5-6] 。
之前经常报道的临床表型严重程度与MCT 8蛋白变异后剩余的转运能力有关。一般认为SLC16A2基因大片段缺失导致MCT 8完全失活而导致严重的表型。虽然SLC16A2基因最常见的大片段缺失是外显子1,但也有报道外显子2-4,外显子2-6,外显子3,外显子3-4和外显子6的缺失。一些SLC16A2致病性错义变异和框内单氨基酸缺失与大量的残留MCT8甲状腺激素转运能力和较轻的临床表型相关,包括一些语言的发育、一些读/写能力和/或在有或无支持的情况下行走的能力[7]。
因AHDS为X染色体隐性遗传,故发病者多累及男性。多数的女性携带者因为X染色体的非随机的失活致无症状或仅仅表现为轻度的甲状腺功能的异常,但无神经系统的受损症状,本例患儿的母亲即无神经系统的症状而仅为甲状腺功能异常,支持文献报道。
AHDS的临床表现可有较大的异质性。肌张力低下、进食困难和早期的体质量增加不足可能出现在生命的最初几周或几个月,也有新生儿黄疸延长的报道。症状轻者可保留一定的运动及语言能力,但仍存在严重的认知障碍;症状重者表现为严重精神运动发育迟缓及无言语能力或言语能力严重受损、先天性肌张力低下、肌张力障碍与锥体束征阳性,对头的控制非常差,不能维持抗重力的姿势[3]。
甲状腺激素异常是AHDS临床诊断的另一个关键发现,但AHDS患者的甲状腺功能异常模式各异。尽管T3水平升高被列为必要的发现,但患者表现为全身肌张力降低和发育迟缓,而不考虑甲状腺功能异常时,应将AHDS作为候选诊断。
AHDS目前尚没有有效的治疗。临床医师可能会把特征性的甲状腺功能异常看作甲状腺功能减退而予以补充左旋甲状腺素或联合丙硫氧嘧啶治疗后,虽患儿机体外周器官的甲状腺素的毒性症状有所改善,但其神经系统的症状并没有明显的减轻,主要原因在于TH的转运障碍。相反有研究表明儿童时期的甲状腺激素替代疗法没有任何益处,且甲状腺功能障碍的恶化可能是危险的[8],且增加家庭经济负担。尚有研究表明不凭借MCT 8蛋白的转运的甲状腺素类似物对 AHDS进行较长时间的治疗随访,提示二碘代丙酸可减轻外周的甲状腺功能的异常及外周甲亢的症状,且无明显的不良反应,但不能减轻患儿的神经系统的损害症状[9]。
研究显示,AHDS的神经系统的损害或许在胎儿的形成过程中即开始出现[10],在神经系统的发育关键期,剥夺TH对神经系统的损害或许是不可逆的,因而此时DITPA的治疗已晚。未经治疗的先天性甲减患儿的临床症状要比AHDS临床症状轻很多的原因也在于此。AHDS的基因治疗目前业已取得一些进步,其应用的前景同样的值得关注[11]。
尽管依据ACMG和AMP的基因序列变异的解释标准和指南 进行致病性的分析显示,还不能确认SLC16A2基因c.193delC半合子变异位点的致病性,但本例患儿有AHDS较为明显的临床表现及特征性的甲状腺功能异常,且c.193delC变异不属于多态性的变化,在人群中出现的概率极其的低下(参考的数据库为1000 Genomes及dbSNP)。故综合患儿的基因结果及临床的表型和甲状腺功能等的信息,可基本确认c.193delC半合子变异位点为患儿的致病性变异。
综上,本例患儿经全外显子基因检测最终诊断为AHDS,发现未见报道的SLC16A2基因c.193delC半合子变异,扩充了AHD的基因变异谱。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
中国生殖健康(2020年4期)2021-01-18 02:58:10
中国生殖健康(2018年4期)2018-11-06 07:12:16
中国现代神经疾病杂志(2017年1期)2017-03-29 06:39:34
湖南畜牧兽医(2016年3期)2016-06-05 08:37:56
兽医导刊(2016年12期)2016-05-17 03:51:42
广东药科大学学报(2016年6期)2016-03-10 07:33:32
放射学实践(2015年2期)2015-02-14 05:38:58
湖北农业科学(2014年11期)2014-09-10 18:06:07
中国神经免疫学和神经病学杂志(2014年5期)2014-05-08 06:17:25
