
Exploring the key genes and pathways of ulcerative colitis in a mouse model using gene expression profiling
2020-12-22 10:42JiaJunWangJianWangXianJuanYangYinFuLiYingWang
Precision Medicine Research 2020年4期
Jia-Jun Wang, Jian Wang*, Xian-Juan Yang, Yin Fu, Li-Ying Wang
Exploring the key genes and pathways of ulcerative colitis in a mouse model using gene expression profiling
Jia-Jun Wang1, Jian Wang1*, Xian-Juan Yang1, Yin Fu1, Li-Ying Wang1
1School of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
Ulcerative colitis is a chronic, relapsing inflammatory disorder of the GI tract. At present, there is no cure for ulcerative colitis. The current treatments only provide a temporary reduction of symptoms because the disease involves many causes. Among them, immune response imbalance, cytokine disequilibrium, and environmental factors are the ulcerative colitis's leading causes. In this study, bioinformatics was used to analyze differentially expressed genes in the Gene Expression Omnibus database and determine the hub genes and pathways related to ulcerative colitis's pathogenesis.
: To compare the differentially expressed genes between ulcerative colitis mouse colon tissue and normal mouse colon tissue, screen out hub genes, explore the biological meaning of these genes, and reveal ulcerative colitis's pathogenesis.: Gene expression profiles (GSE22307) were downloaded from the Gene Expression Omnibus database. We conducted differential screens of the expression of genes (differentially expressed genes) between two groups using the online tool Gene Expression Omnibus 2R, then use the ClueGO plugin in Cytoscape software to perform gene ontology and Kyoto Encyclopedia of Genes and Genomes analysis on differentially expressed genes. Molecular Complex Detection plugin was used to visualize differentially expressed genes' gene-gene interaction network and screen for gene modules. The cytohubba plugin was used to screen the hub genes and then analyzed with the ClueGO plugin to further understand ulcerative colitis's specific biological mechanism.: A total of 1,151 differentially expressed genes were identified, among which 798 were upregulated, and the remaining 353 were down-regulated. The gene ontology results revealed that these differentially expressed genes were primarily enriched in inflammation, cytokine activity, protein binding, and several other biological processes. Kyoto Encyclopedia of Genes and Genomes analysis indicated the differentially expressed genes were mainly increased in the TNF and IL-17 signaling pathway. Using the gene-gene interaction network analysis, we found that,,,,, andmay be responsible for ulcerative colitis's occurrence and development. Those target genes were enriched mainly in the neutrophil apoptotic process and positive regulation of humoral immune response.: The results of our study indicated that those differentially expressed genes, especially,,,,, and, may be associated with the development and progression of ulcerative colitis.
Bioinformatics analysis, Ulcerative colitis, Differentially expressed genes, Gene
Background
Ulcerative colitis (UC), a type of inflammatory bowel disease, is a chronic inflammatory disorder of the colon, which lesion location usually begins in the rectum and ends in the terminal ileum [1]. UC was reported as more common in young adults; the victims' mean age is between 30 and 40 years [2]. UC has no gender dominance, and the prevalence of it has been increasing worldwide. UC's pathogenesis is complex and involves both immunological and genetic factors. It can cause diarrhea, abdominal pain, and bloody stool, seriously affecting patients' quality of life [3]. Although the medical community has discussed a lot about UC's etiology and pathogenesis, no consensus has been reached yet.
The pathogenesis of UC mainly involves the changes in the intestinal mucosa and intestinal epithelial cells. In recent years, molecular biology and genomics play an essential role in explaining the pathophysiology of diseases. Thus far, genome-wide association studies have identified more than 240 susceptibility gene loci for UC, such as,,, et al. [4] In the present study, GSE22307 was obtained from the Gene Expression Omnibus (GEO) database and analyzed by bioinformatics to determine UC's possible molecular mechanisms.
Materials and methods
Gene expression microarray data
Gene expression profile GSE22307 comes from the GEO database (www.ncbi.nlm.nih.gov/geo/)[5]. C57BL/6 mice received 3% dextran sodium sulfate (DSS) in the drinking water to induce colitis, and collect colon tissues from each cohort on days 0 and 6. Total RNA is extracted from colon tissue and detected by Affymetrix GeneChip Mouse Genome 430 2.0 Array. In this study, mice modeled with 0-day DSS were taken as the normal group (sample number is 5) and mice modeled with DSS on day 6 were UC group (sample number was 6).
Differentially expressed genes in UC colon tissue
The original data files used for analysis include TXT files (Affymetrix platform). GEO2R (ncbi.nlm.nih.gov/geo/geo2r/) was used to analyze the differentially expressed genes (DEGs) between the normal and the UC groups. The absolute value of log fold-change > 1 and an adjusted-value < 0.05 were defined as the filter condition. The DEGs with statistical significance between the two groups were selected and identified.
Gene ontology and Kyoto Encyclopedia of Genes and Genomes analysis of DEGs
Functional enrichment analysis was performed using Cytoscape software [6] (version 3.8.0) (www.Cytoscape.org) and the ClueGO plugin [7] (version 2.57, Laboratory of Integrative Cancer Immunology, Bethesda, MD, USA). Gene ontology (GO) analysis was used to predict the potential functions of DEGs in biological processes (BP), molecular functions, and cellular components. The pathway analysis was performed on the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. It is considered that the representative pathway category with a-value < 0.05 is statistically significant.
Construction of the gene-gene interaction network
A large number of DEGs we obtained are potential UC-related genes; this suggests that these DEGs in colon tissue may be involved in the development of UC. First, the gene-gene interaction network was searched using the STRING (search tool for recurring instances of neighboring genes) tool [8] (http://www.STRING-db.org/)and the combined score > 0.4 was saved and exported. Subsequently, Cytoscape software (version 3.8.0) was used to visualize the UC genes' interaction regulatory network. Then, the plugin MCODE (molecular complex detection) [9] was used to screen the two most important sub-network modules of the gene interaction network in Cytoscape. Sub-module establishment standard: degree cutoff = 2, k-core = 2, maximum depth = 100, node density cutoff = 0.1
Screening and analysis of hub genes
The 5 built-in Cytohubba plugin [10] methods (degree, betweenness, EPC, MCC, and stress) were used to score all DEGs on the gene-gene interaction regulatory network and screened the top 20 DEGs for each scoring method. Then the VennPainter [11] software was used to fet alh overlapping genes, and the result was the hub gene. Finally, we used the ClueGO plugin to perform GO and KEGG enrichment analysis of the hub genes to determine further the biological significance and pathways related to UC.
Results
Identification of DEGs
Gene expression data for UC was downloaded from the GEO, GEO2R was used to identify DEGs between UC colon tissue samples and normal controls. In total, 1,151 DEGs were identified, including 798 upregulated and 353 downregulated genes. The volcano plot and heatmap of DEGs are shown in Figure 1. Table 1 shows the top 10 upregulated and the top 10 downregulated genes' specific information of DEGs.
GO enrichment analysis of DEGs
GO analysis of DEGs was performed using the ClueGO in Cytoscape software. GO enrichment analysis (Figure 2) indicated that these DEGs were significantly enriched in the inflammatory response, immune system process, autoimmune response, neutrophil chemotaxis, and other BP. For molecular functions, DEGs were mainly enriched in cytokine activity, chemokine activity, peptidase activity, and protein binding. Besides, GO cellular components analysis showed that DEGs were significantly enriched outside the cell, on the cell surface, and outside the plasma membrane.
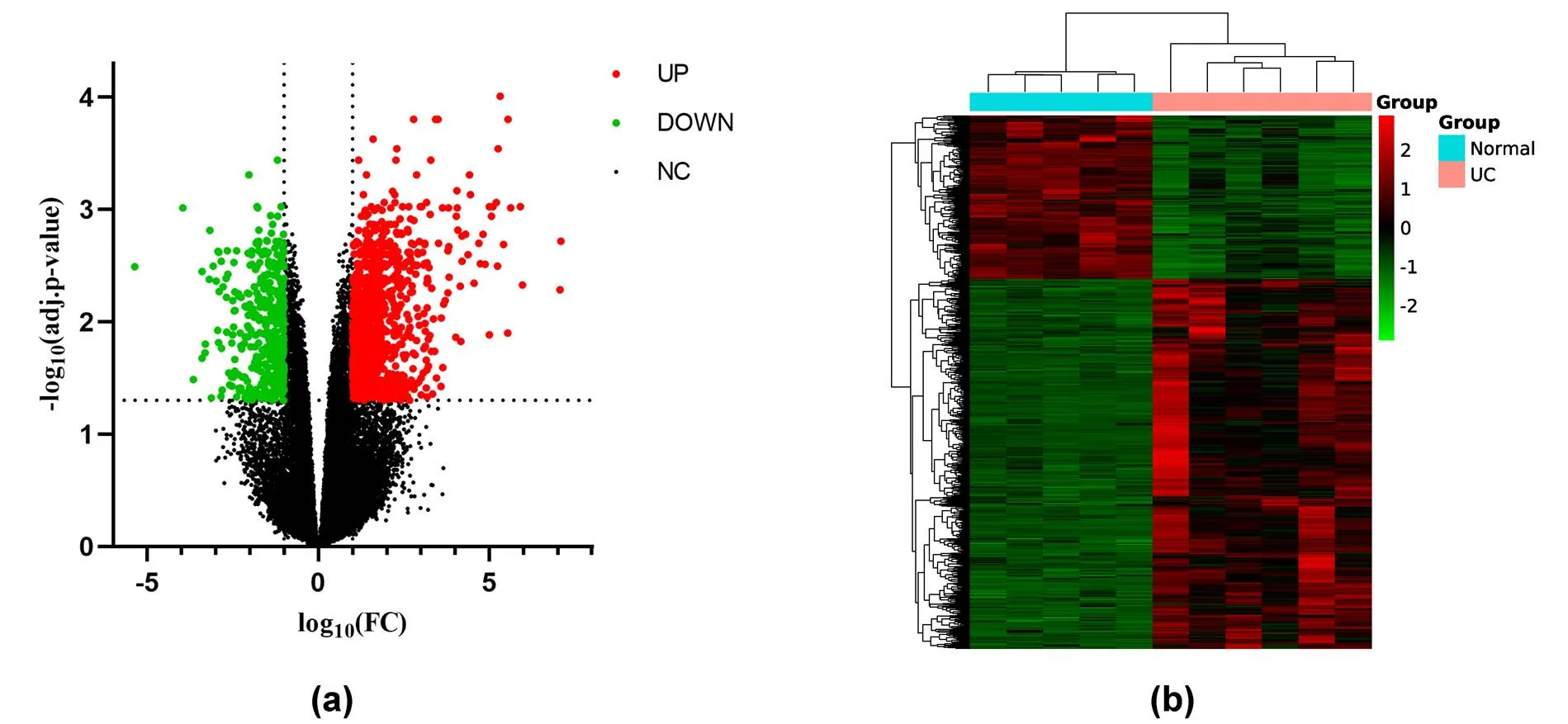
Figure 1 The expression changes of all genes and the expression heat map of DEGs. (a) The volcano plot shows the expression changes of all genes. Red indicates significantly upregulated DEGs. Green indicates significantly down-regulated DEGs, and black indicates genes that are not significantly changed. (b) The heat map shows all DEGs that have changed considerably. Red indicates upregulated DEGs substantially, and green shows significantly down-regulated DEGs. DEGs, differentially expressed genes.
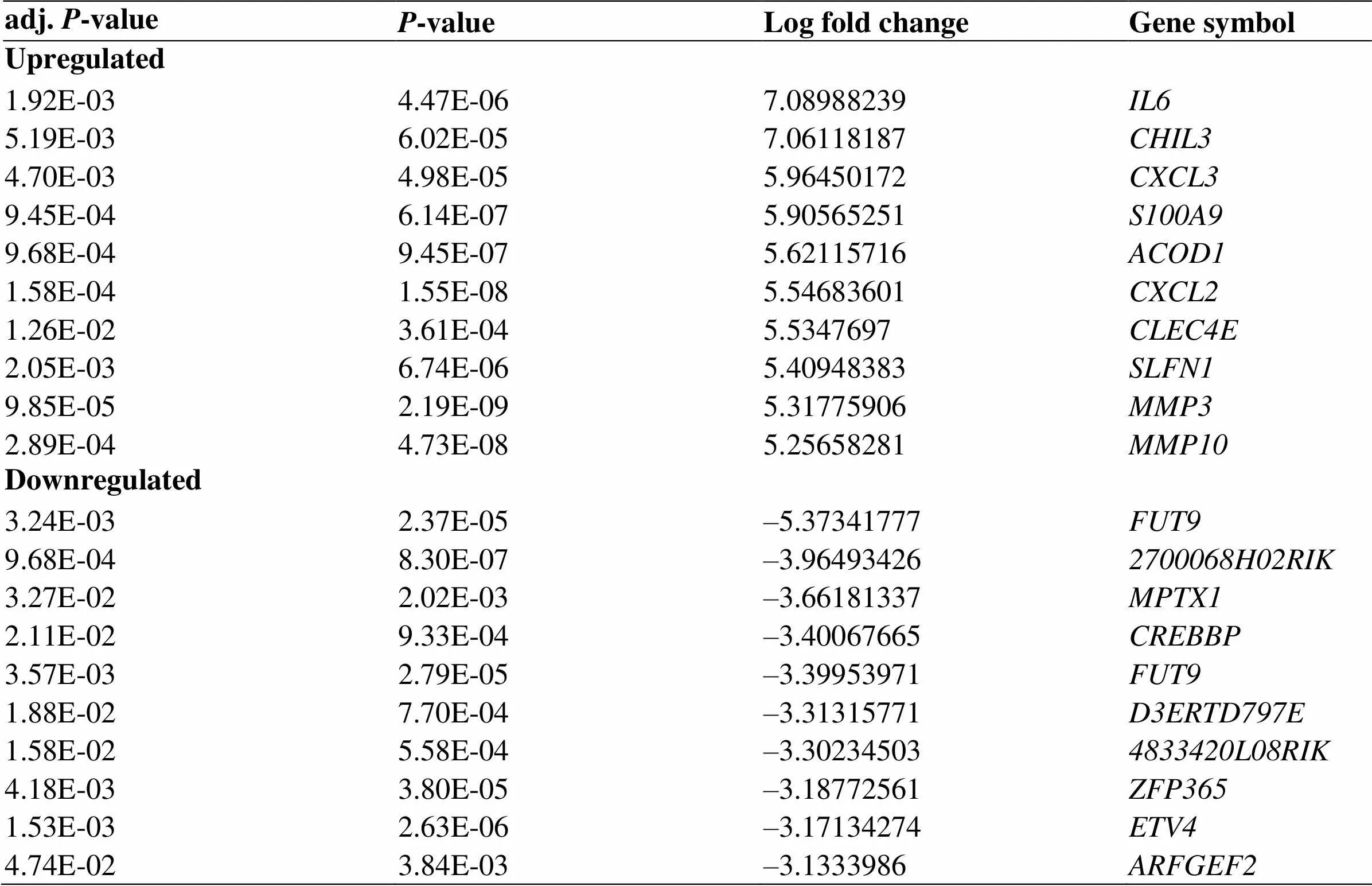
Table 1 The top 10 regulated differentially expressed genes between the ulcerative colitis colon tissue and the normal colon tissue
KEGG enrichment analysis of DEGs
The KEGG analysis results indicated that the target genes were riched in the malaria,IL-17 signaling pathway, TNF signaling pathway, AGE-RAGE signaling pathway in diabetic complications, toll-like receptor signaling pathway, chemokine signaling pathway, JAK-STAT signaling pathway, et al. (Figure 3 and Table 2).
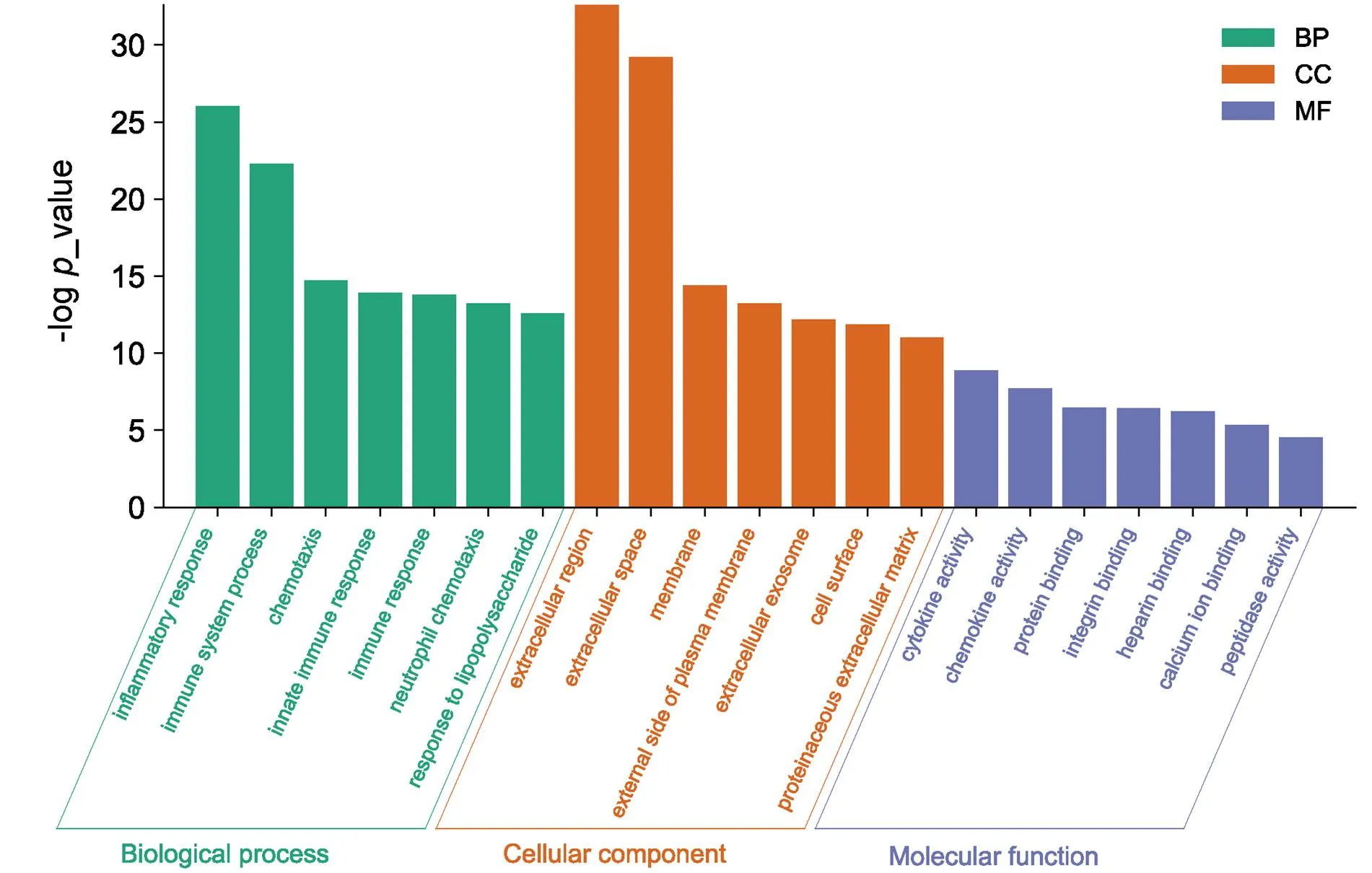
Figure 2 GO analysis of DEGs. Green represents biological processes, orange represents cellular components, and purple represents molecular functions. DEGs, differentially expressed genes; GO, gene ontology;CC, cellular component; MF, molecular function; BP, biological process.
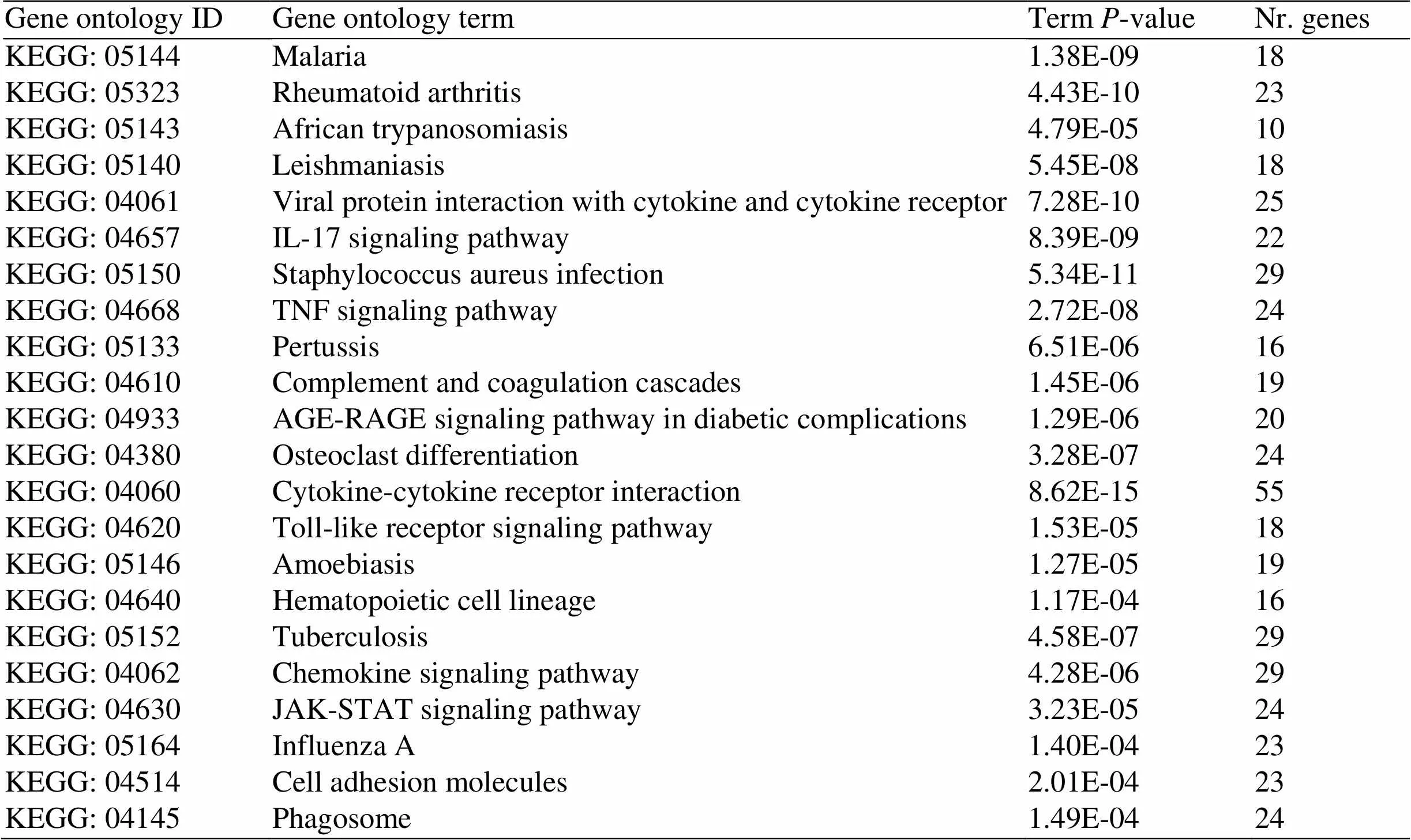
Table 2 Core pathways
KEGG, Kyoto Encyclopedia of Genes and Genomes.
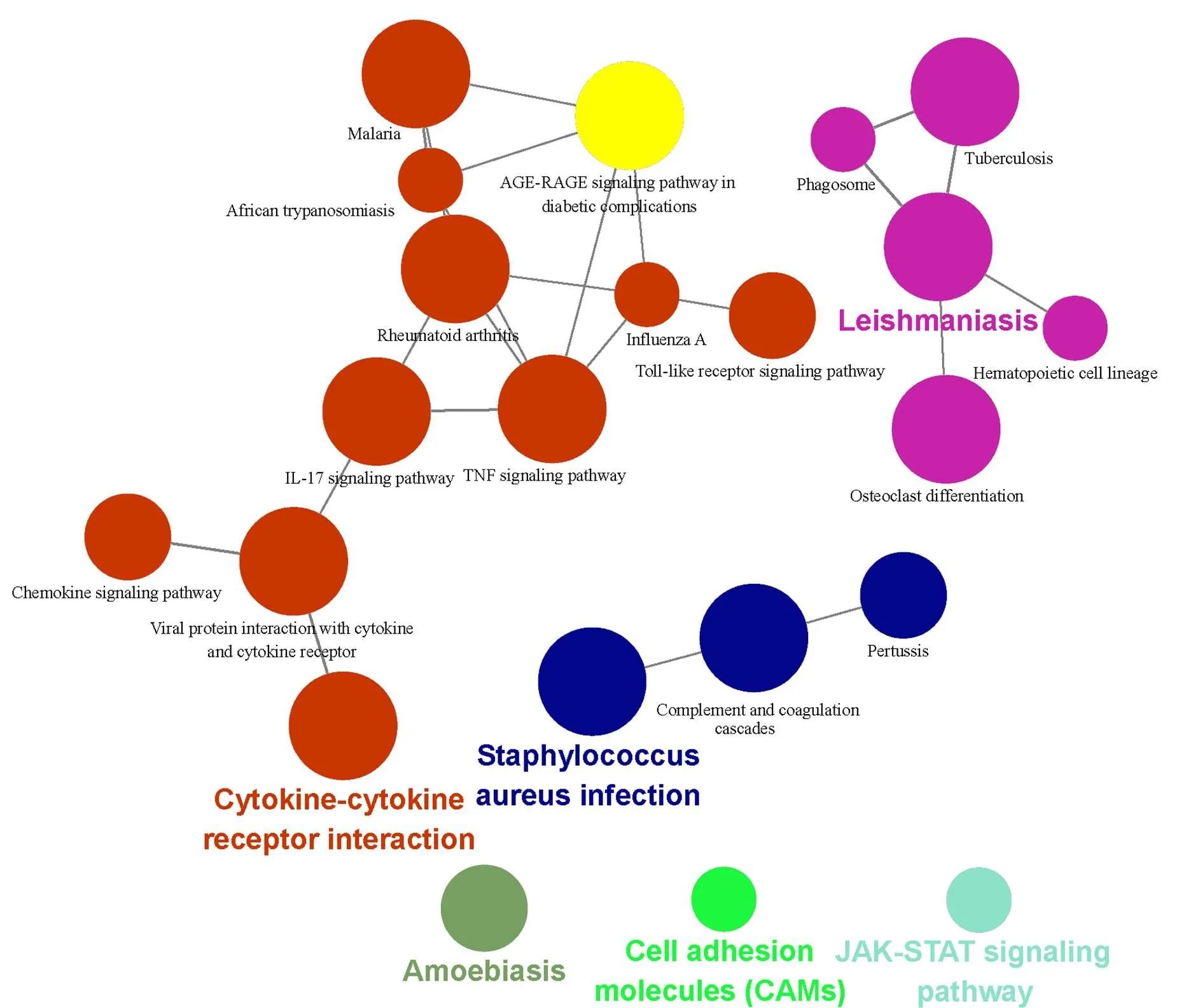
Figure 3 KEGG enrichment analysis of DEGs. Different node colors mean different pathways. The darker the color indicates the more enriched the results are, and the closer the colors are, the tighter the pathways are clustered. DEGs, differentially expressed genes; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Gene interaction network construction
According to the STRING database's information, the inclusion standard of gene interaction network was a total score > 0.4, A total of 1,085 nodes, and 9,007 edges were obtained.
Nodes represent genes, and edges represent regulatory relationships between genes and genes. The networks were visualized using the Cytoscape software; then, the significant modules were identified via the MCODE plugin (Figure 4). The MCODE score of module "a" was 36.098, consisting of 83 nodes and 1,480 edges, and the MCODE score of module "b" was 12.421, consisting of 58 nodes and 354 edges
Screening and enrichment analysis of hub genes
Using the cytoHubba plugin to select the hub gene, hub genes were identified according to the degree, betweenness, EPC, MCC and stress algorithms (Table 3). Then screened out overlapping genes through VennPainter software; a total of 6 hub genes were identified (Figure 5). Finally, the ClueGO plugin was used to perform GO and KEGG enrichment analysis on these 6 hub genes, and the results were shown in Table 4.

Table 3 Five algorithms to identify the top 20 hub genes
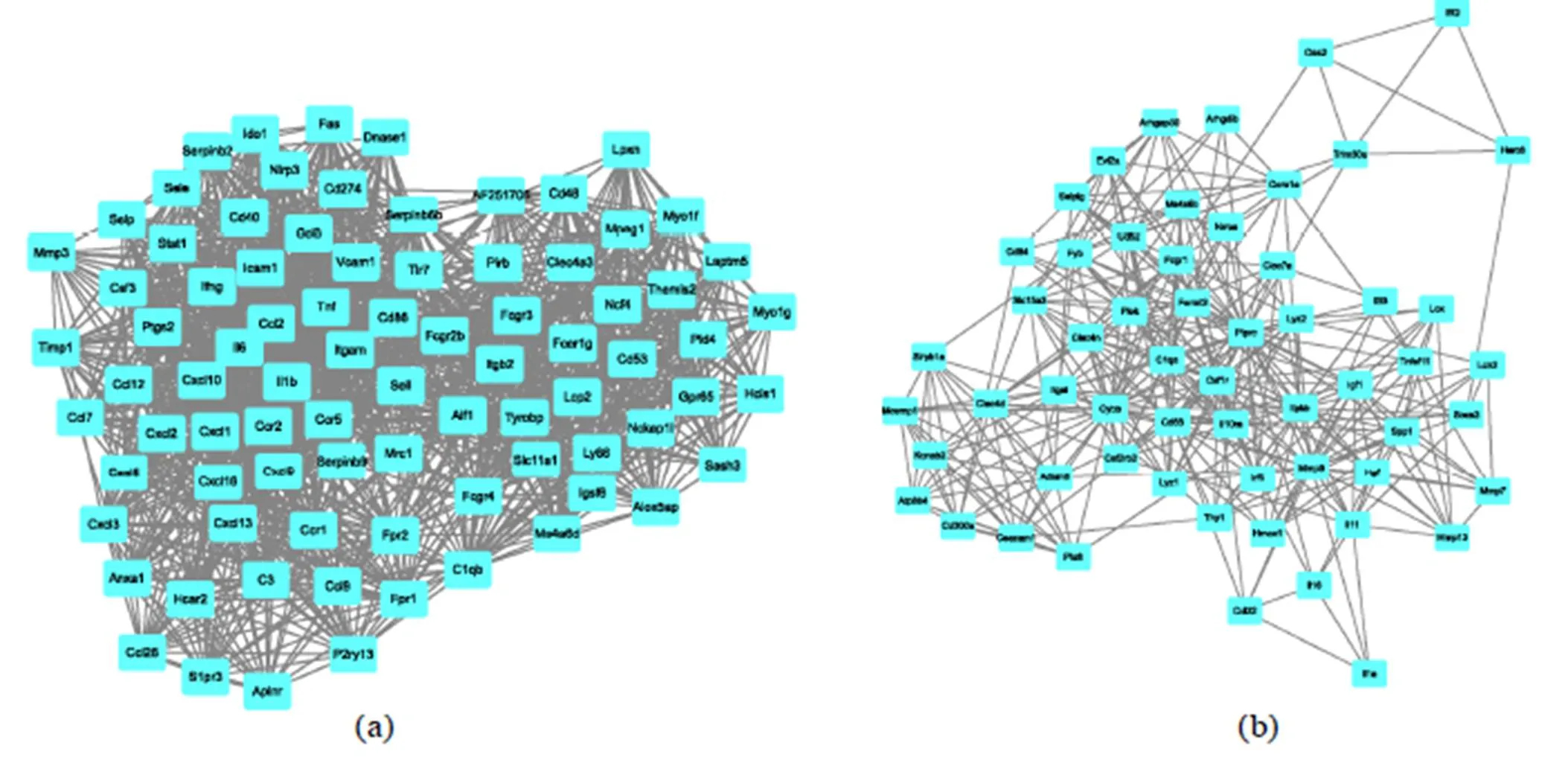
Figure 4 The top two sub-modules of the gene interaction network. The rectangles represent the genes in the module, while the lines represent the interaction between genes.
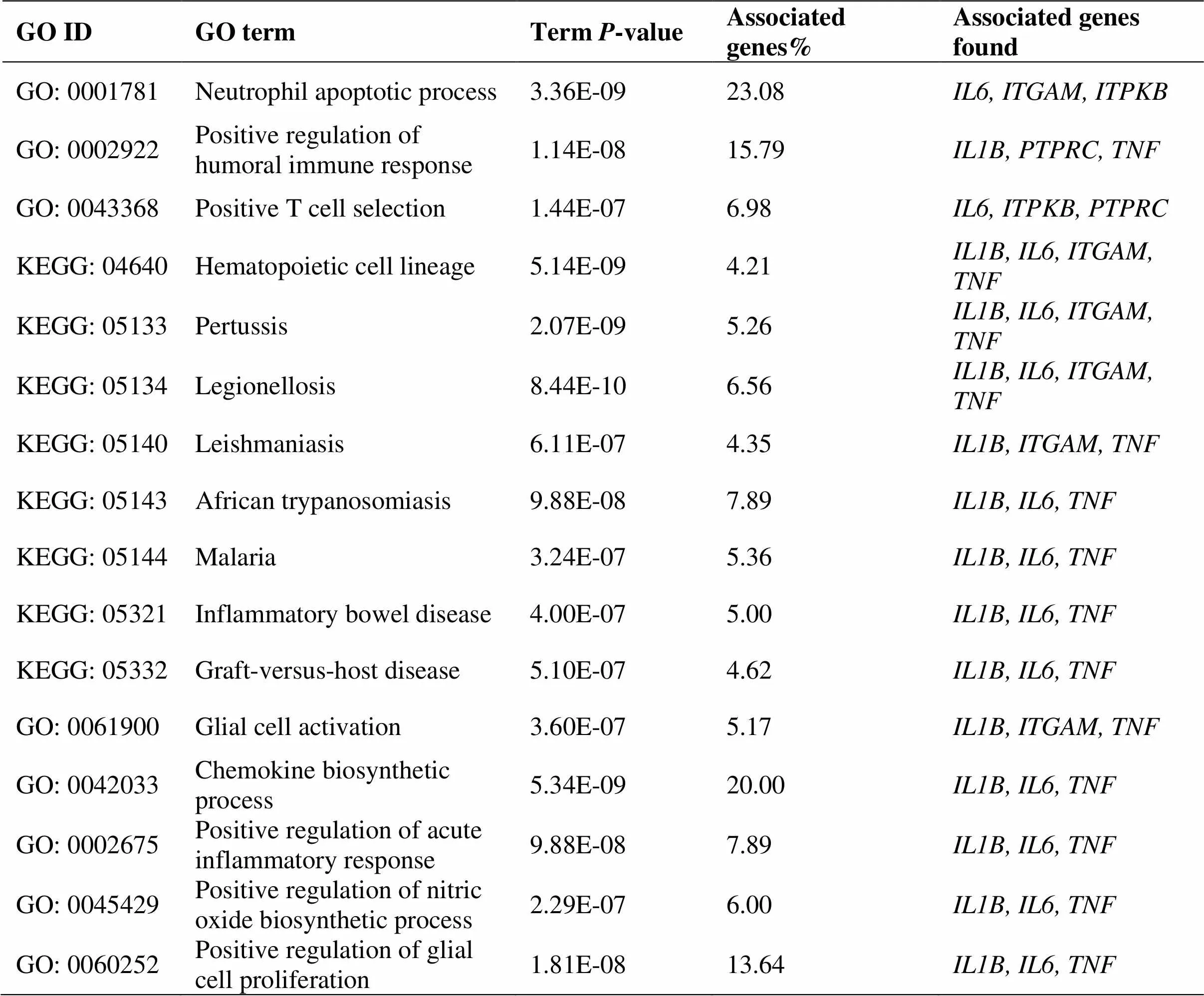
Table 4 GO and KEGG enrichment analysis of hub genes
KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, gene ontology.
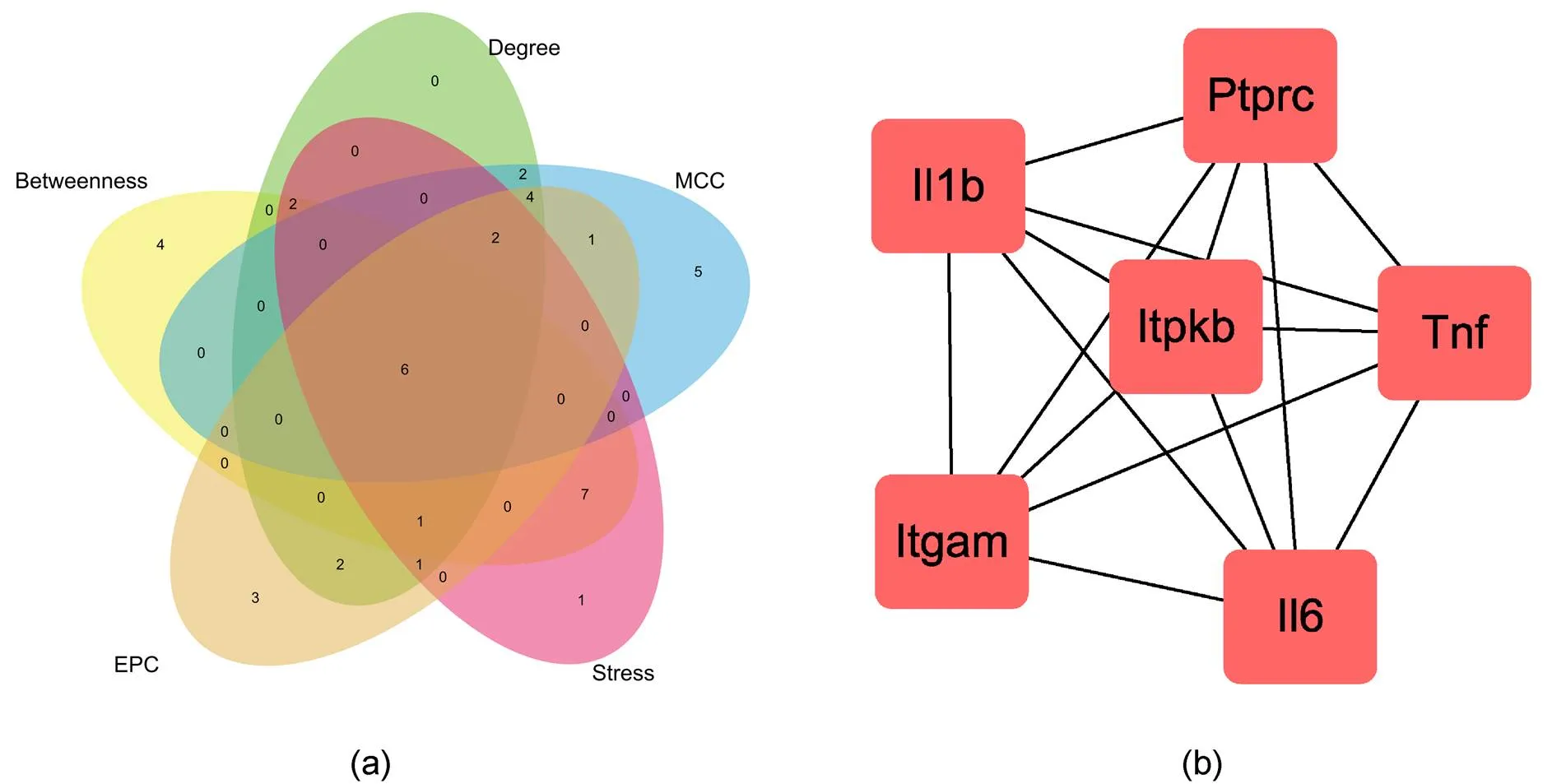
Figure 5 (a) The overlapping genes in the five algorithms were identified by VennPaint software. (b) Hub genes interaction network.
Discussion
UC is an easily relapsing and refractory autoimmune disease, which characteristics include the destruction of the intestinal barrier, imbalance of immune response, and cytokines. Among these, the intestinal barrier's collapse is the direct cause of UC's occurrence and development [12, 13, 14]. In this study, we downloaded the gene expression profile of GSE22307 and performed bioinformatics analysis. The results showed that compared with the normal control group, there were 1,151 DEGs in the UC colon tissue. Besides, GO, KEGG pathways, and the gene-gene interaction network analysis was performed to identify biomarkers or significant genes related to UC.
We performed GO functional terminology and KEGG pathway enrichment analysis on DEGs' hub genes to clarify the fundamental difference in molecular mechanisms between UC colon tissue and normal colon tissue. Consider the results of GO analysis, and we associated the hub genes with the following BP: cytokine activity, autoimmune response, and neutrophil chemotaxis, which may have an intimate association with UC. KEGG pathway enrichment results showed that TNF signaling pathway, IL-17 signaling pathway, and JAK-STAT signaling pathway are most related to UC. This is similar to the previous report. cHANG SU et al. [15]. Added 5% DSS to drinking water for seven days to establish a UC mouse experimental model; they found that the IL-17 and Th17 cell content in the model mice's colon tissue was significantly increased. STAT1 and STAT3 transcription factors can mediate the proliferation and survival of IL-6; cocoa extract was discovered to target the JAK-STAT signaling pathway in UC by decreasing the expression of[16, 17]. The KEGG analysis results also indicated that UC's molecular mechanism might be related to Pertussis, malaria, and African trypanosomiasis, et al. which had not been seen in previous reports.
It is worth noting that there was abundant evidence of DEGs in UC colon tissue that have been proven to play essential roles during UC's pathological process. We used the 5 algorithms of Cytoscape software to analyze the top 20 critical nodes in the STRING database; it was finally found that there were six hub genes, including,,,,, and. Immune hyperactivity and inflammatory cell infiltration were the leading causes of intestinal barrier destruction. Zhou Qing et al [18]found that the TNF-α/IL-6/GP130 inflammatory immune axis is closely related to the expression of colonic inflammatory factors and immune hyperactivity in UC mouse models. XUESONG BAI et al. [19] found that the serum levels of MMP-2, MMP-9, and IL-6 in patients with active UC were significantly increased. Xinhua Li et al. [20] showed that miR-219-5p plays a vital role in the pathogenesis of UC, growing Treg cells' percentage by upregulatingand decrease the rate of Th17 cells by down-regulatingand. Besides, John H et al. [21]found thatgene defects weaken the chemotaxis and adhesion of neutrophils and T cells to the endothelium stimulated by VEGF-A. In short,,,,,,,, andhave been reported to participate in UC's development.
Similar bioinformatics analysis of rat models or human colon tissue helps to further reveal essential genes in UC. Xiong Di et al. [22]found that,, C1R,, andmay be potential target genes for the diagnosis and treatment of UC by using the GSE65114 gene chip. Qiu Shou et al. [23] analyzed the DEGs of the gene expression profile GSE16879 through the weighted gene co-expression network and screened the high-risk driver genes of UC, including,,,,,,,, andandare also reported in our study. Hu Zong Ren et al. [24] analyzed the GSE36807 gene chip, and the results showed that,,,,,are potentially essential genes related to the pathogenesis of UC.family genes are a class of chemokines, which can induce specific cells such as T cells and neutrophils to accumulate at specific sites, helping inflammation and ulcers occur.
For the BP involved in the pathogenesis of UC, GO analysis results showed that it is related to inflammatory response, immune system process, response to lipopolysaccharide, and neutrophil chemotaxis. The imbalance of inflammatory factor expression is the direct factor leading to UC. Current treatment strategies mainly focus on suppressing or regulating excessive long-standing immune responses and the potential reversal of mucosal inflammation [25, 26]. For the cell components in the GO analysis results, it showed that DEGs are mainly enriched on the surface and outside of the cell membrane, changes in the intestinal epithelial cell membrane composition will change its permeability, leading to damage to the intestinal epithelial barrier and causing bacteria and other antigens to invade the intestine, then trig immune response [27–29]. Several studies have shown that impaired intestinal barrier is an essential cause of UC, which is consistent with our analysis results [30–32]. In terms of the molecular function of DEGs, we found that they mainly affect the activity of cytokines, chemokines, and protein binding. In UC, chemokines induce cytokines to accumulate at inflammation sites, cytokines mediate inflammation, and protein binding is related to the tight junctions of intestinal epithelial cells [33–36].
In summary, 1,151 DEGs were identified, including 798 upregulated DEGs and 353 downregulated DEGs, between UC colon tissue and normal colon tissue in a mouse model. GO and KEGG pathway analysis provided a series of related hub genes and pathways to understanding UC colon tissue lesions' molecular mechanisms. The data suggest that,,,,, andgenes are positively correlated with UC's colon tissue development.
1. Ungaro R, Mehandru S, Allen PB, et al. Ulcerative colitis. The Lancet 2017, 389: 1756–1770.
2. Gan D, Han CP. Research progress on the treatment of ulcerative colitis with integrated traditional Chinese and western medicine. Mod J Integr Tradit Chin West Med 2017, 26: 2958–2961.
3. Silva BC da, Lyra AC, Rocha R, et al. Epidemiology, demographic characteristics and prognostic predictors of ulcerative colitis. World J Gastroenterol 2014, 20: 9458–9467.
4. Kakuta Y, Ichikawa R, Fuyuno Y, et al. An integrated genomic and transcriptomic analysis reveals candidates of susceptibility genes for crohn's disease in Japanese populations. Sci Rep London 2020, 10: 10236.
5. Lan K, Wang D, Fong S, et al. A survey of data mining and deep learning in bioinformatics. J Med Syst 2018, 42: 139.
6. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13: 2498–2504.
7. Gabriela B, Bernhard M, Hubert H, et al. ClueGO: a Cytoscape plugin to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25: 1091–1093.
8. Szklarczyk D, Morris J H, Cook H, et al. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res 2017, 45: D362–D368.
9. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformat 2003, 21: 27–29.
10. Chin CH, Chen SH, Wu HH, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol 2014, 8: S11.
11. Lin G, Chai J, Yuan S, et al. VennPainter: a tool for the comparison and identification of candidate genes based on venn diagrams. PLOS ONE 2016, 11: e0154315.
12. Gajendran M, Loganathan P, Jimenez G, et al. A comprehensive review and update on ulcerative colitis. Dis Month 2019, 65: 100851.
13. Eisenstein M. Ulcerative colitis: towards remission. Nature 2018, 563: S33.
14. Neurath MF, Leppkes M. Resolution of ulcerative colitis. Semin Immunopathol 2019, 41: 747–756.
15. Su C, Liu S, Ma X, et al. Decitabine attenuates dextran sodium sulfate-induced ulcerative colitis through regulation of immune regulatory cells and intestinal barrier. Int J Mol Med 2020, 46: 583–594.
16. Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell, Cambridge: Cell Press 2009, 15: 103–113.
17. Andlujar I, Carmen Recio M, Giner R M, et al. Inhibition of ulcerative colitis in mice after oral administration of a polyphenol-enriched cocoa extract is mediated by the inhibition of STAT1 and STAT3 phosphorylation in colon cells. J Agric Food Chem 2011, 59: 6474–6483.
18. Zhou Q, Chen YG, Shang HT, et al. Based on the TNF-α/IL-6/GP130 inflammatory immune axis, the study of Huangkui Lianchang decoction on inflammatory factors and mucosal inflammation immune hyperactivity in mice with ulcerative colitis. Lishizhen Med Mater Med Res 2019, 30: 2833–2837.
19. Bai X, Bai G, Tang L, et al. Changes in MMP-2, MMP-9, inflammation, blood coagulation and intestinal mucosal permeability in patients with active ulcerative colitis. Exp Ther Med 2020, 20: 269–274.
20. Li X, Sun L, Chen L, et al. Upregulation of microRNA-219-5p relieves ulcerative colitis through balancing the differentiation of Treg/Th17 cells. Eur J Gastroen Hepat 2020, 32: 813–820.
21. Chidlow JH, Glawe JD, Alexander JS, et al. VEGF (164) differentially regulates neutrophil and T cell adhesion through ItgaL- and ItgaM-dependent mechanisms. Am J Physiol Gastr L 2010, 299: G1361–G1367.
22. Xiong D, He W, Li Y, et al. Bioinformatics analysis of gene expression profiling in colonic mucosa of patients with ulcerative colitis. Med Pharmaceut J Chin People's Liber Army 2019, 31: 50–55.
23. Qiu SH, Zhang KX, Ren DD, et al. Weighted gene co-expression network analysis of pathogenesis of ulcerative colitis. J Mudanjiang Med Univ 2020, 41: 49–55.
24. Hu ZHR, Zhen WJ, Yan Q, et al. Bioinformatic analysis of differentially expressed genes and Chinese medicine prediction for ulcerative colitis. China J Chin Mater Med 2020, 45: 1684–1690.
25. Ahluwalia B, Moraes L, Magnusson MK, et al. Immunopathogenesis of inflammatory bowel disease and mechanisms of biological therapies. Scand J Gastroentero 2018, 53: 379–389.
26. Yao D, Dong M, Dai C, et al. Inflammation and inflammatory Cytokine contribute to the initiation and development of ulcerative colitis and its associated cancer. Inflamm Bowel Dis 2019, 25: 1595–1602.
27. Camilleri M, Madsen K, Spiller R, et al. Intestinal barrier function in health and gastrointestinal disease. Neurogastroent Motil 2012, 24: 503–512.
28. Peterson L W, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 2014, 14: 141–153.
29. Wu Y, Tang L, Wang B, et al. The role of autophagy in maintaining intestinal mucosal barrier. Am J Physiol-Lung C 2019, 234: 19406–19419.
30. Kielland A, Blom T, Nandakumar KS, et al. In vivo imaging of reactive oxygen and nitrogen species in inflammation using the luminescent probe L-012. Free Radical Bio Med 2009, 47: 760–766.
31. Hong H S, Hwang D Y, Park J H, et al. Substance-P alleviates dextran sulfate sodium-induced intestinal damage by suppressing inflammation through enrichment of M2 macrophages and regulatory T cells. Cytokine 2017, 90: 21–30.
32. Ko J-A, Yanai R, Nishida T. Up-regulation of ZO-1 expression and barrier function in cultured human corneal epithelial cells by substance P. FEBS Letters 2009, 583: 2148–2153.
33. Wang J, Zhang C, Guo C, et al. Chitosan ameliorates DSS-induced ulcerative colitis mice by enhancing intestinal barrier function and improving microflora. Int J Mol Sci 2019, 20: 21–32
34. Tan Y, Guan Y, Sun Y, et al. Correlation of intestinal mucosal healing and tight junction protein expression in ulcerative colitis patients. Am J Med Sci 2019, 357: 195–204.
35. Xue B, Lui XL, Dong WW, et al. EGCG maintains Th1/Th2 balance and mitigates ulcerative colitis induced by dextran sulfate sodium through TLR4/MyD88/NF-κB signaling pathway in rats. Eur J Gastroen Hepat 2017, 30: 23-33.
36. Vinayaga-Pavan M, Frampton M, Pontikos N, et al. Elevation in cell cycle and protein metabolism gene transcription in inactive colonic tissue from icelandic patients with ulcerative colitis. Inflamm Bowel Dis 2019, 25: 317–327.
UC, ulcerative colitis; DEGs, differentially expressed genes; GEO, Gene Expression Omnibus; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DSS, dextran sulfate sodium salt; BP, biological process; STRING, the search tool for recurring instances of neighboring genes; MCODE, molecular complex detection.
The authors declare that they have no conflict of interest.
: 08 December 2020
*Corresponding to: Jian Wang. Chengdu University of Traditional Chinese Medicine,1166 Liutai Avenue, Wenjiang District, Chengdu 611137, China. E-mail: jianwang08@163.com.
:
Jia-Jun Wang, Jian Wang, Xian-Juan Yang, et al. Exploring the key genes and pathways of ulcerative colitis in a mouse model using gene expression profiling. Precision Medicine Research 2020, 2 (4): 143–152.
: Xiao-Hong Sheng.
: 25 August 2020,
04 December 2020,
