
不同势能模型下的水分子动力学性质
2020-12-21 03:58俞联梦
工业技术创新 2020年5期
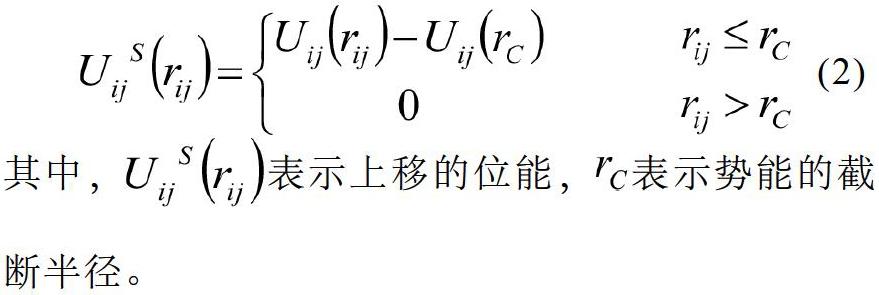

摘 要: 采用分子动力学方法,对SPC、SPCE、TIP3P、TIP4P、TIP5P五种势能模型下水分子的热力学性质、径向分布函数、自扩散系数进行计算,优选最适合于水分子动力学性质研究的势能模型。研究发现:1)采用SPCE模型通过模拟得到的内能、汽化焓最为准确,其他模型受到了模型固有性质和系综(大量宏观上完全相同的体系的抽象集合)的影响,模拟结果不甚理想;2)采用SPCE、TIP4P、TIP5P模型可以较好地反映水分子短程有序、长程无序的微观结构;3)采用SPCE模型通过模拟得到的自扩散系数相对误差仅为0.39%,远好于其他模型。结果表明,SPCE模型能够较真实地反映水分子的热力学性质、微观结构和自扩散特性等动力学性质。
关键词: 水分子;势能模型;动力学;径向分布函数;自扩散系数
中图分类号:O645 文献标识码:A 文章编号:2095-8412 (2020) 05-085-05
工业技术创新 URL: http://gyjs.cbpt.cnki.net DOI: 10.14103/j.issn.2095-8412.2020.05.016
引言
水是生命的源泉,水分子的研究對水的开发利用具有非常重要的意义。分子动力学(MD)方法是模拟水分子动力学性质的一种非常有效的方法,一直受到国内外研究者的关注[1-3]。
单个水分子看似结构比较简单,但氢键的作用使得水分子相互聚合,形成多分子聚合物,水分子的微观结构发生了复杂化。采用分子动力学方法研究水分子,最重要的也是选择分子间的相互作用势。在水分子动力学计算中,可以选择多种势能模型,包括量子力学从头计算模型(如MCY模型[4])、半经验模型(如ST2、SPC、SPCE、TIP3P、TIP4P、TIP5P)等[5-8]。MCY模型结构较为复杂,计算时间成本较高,因此在实际应用中更多采用半经验模型。
本文对水分子的SPC、SPCE、TIP3P、TIP4P、TIP5P等势能模型进行计算,考察水分子的热力学性质、径向分布函数、自扩散系数,优选最适合于水分子动力学性质研究的势能模型。
1 势能模型
SPC、SPCE、TIP3P、TIP4P、TIP5P这几种模型都是刚体固定点电荷模型,计算时选用非极性作用Lennard-Jones势模,分布为有效电荷表征电子云。
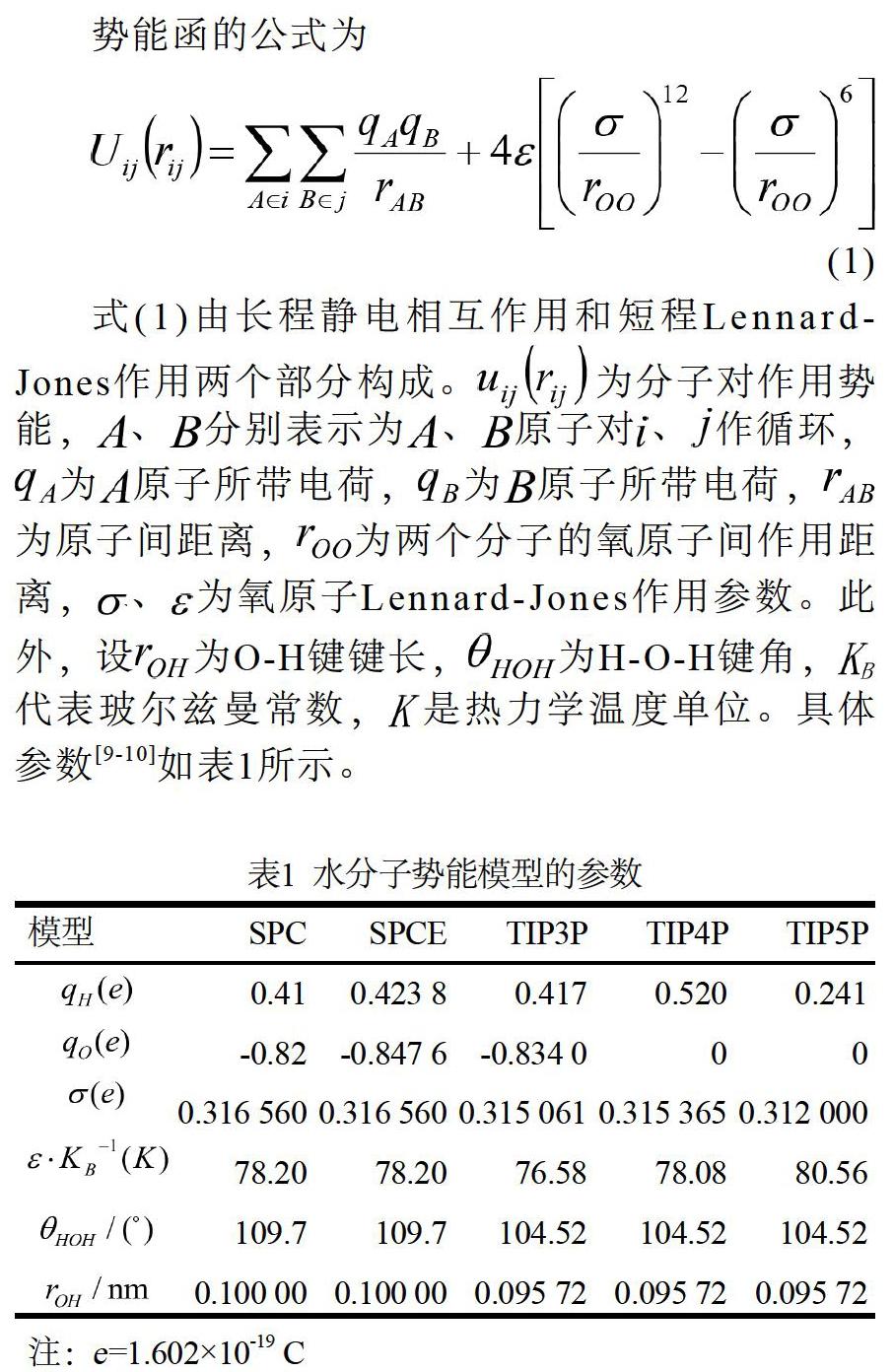
势能函的公式为
式(1)由长程静电相互作用和短程Lennard-Jones作用两个部分构成。为分子对作用势能,、分别表示为、原子对、作循环,为原子所带电荷,为原子所带电荷,为原子间距离,为两个分子的氧原子间作用距离,、为氧原子Lennard-Jones作用参数。此外,设为O-H键键长,为H-O-H键角,代表玻尔兹曼常数,是热力学温度单位。具体参数[9-10]如表1所示。
势能的截断,会使计算时能量在边界处发生断裂,导致哈密顿函数不守恒。采用上移的势能函数[7]来弥补缺陷,计算公式为
其中,表示上移的位能,表示势能的截断半径。
2 模拟方法
模拟的水分子总数为300个,模拟温度为298 K、压强为1 atm(常温常压),模拟系综(系综表示大量宏观上完全相同的体系的抽象集合)为正则系综(NVT),水的起始密度设为1.0 g/cm3。
计算模型设为面心立方晶格,计算开始时水分子取向随机,各分子的起始速度按Maxwell-Boltzmann分布取样,对温度的控制采用速度标定法,以确保体系总动量为零。
计算过程中采用最小镜像准则和两体有效势基本近似,周期性边界条件采用立方结构[11],计算盒子尺寸由密度确定。采用球形截去法,设置截断半径为0.9 nm。采用长程校正法校正截断半径之外的分子间相互作用能,长程静电相互作用采用Ewald加和法[12]进行计算。
几何构型的固定采用SHAKE算法,采用5阶Gear预估—校正法[13]求解运动方程。计算时所取时间步长为2 fs,每次模拟总时间为300 ps,前200 ps使体系达到平衡,后100 ps用于进行统计计算各种物理性质。
3 结果与讨论
3.1 热力学性质
计算常温常压下,不同势能模型下水的内能、汽化焓,并把所得结果与实验值和其他文献值进行比较。
汽化焓计算公式为
其中,H表示焓值,E代表体系内能,P是压强,V是体积,T是温度,R是普适气体常量。
模拟结果与比较如表2所示,其中MC代表量子力学从头计算模型。选用SPCE模型模拟得到的水的内能、汽化焓与实验值和其他文献值较接近,而其他模型可能受到了模型固有性质和系综的影响,模拟结果不太理想。总之,采用SPCE模型计算水分子的热力学性质最为合适。
3.2 径向分布函数
径向分布函数是反映流体微观结构的物理量,它表明了与某个原子距离为的单位体积元内出现的分子数,表达式为
其中,为体系的体积,为分子数,为两个分子的质心的距离,为函数。
计算常温常压下水分子在不同势能模型下的gOO(O-O径向分布)、gHO(H-O径向分布)、gHH(H-H径向分布),并与实验值[12]进行比较。
图1a所示为O-O径向分布函数与实验值。可以看出,不同势能模型的O-O径向分布函数与实验值的峰值变化趋势都相符,其中SPCE、TIP4P和TIP5P三个模型与实验值一样出现了三个峰值,SPC、TIP3P只有一个峰值。综合来看,在研究水的O-O径向分布函数时,选用TIP5P模型更为合适。此外,SPCE、TIP4P和TIP5P模型的第二峰值出现在0.45 nm,第三峰值出现在0.68 nm,这与文献[15]使用蒙特卡罗方法所得结果相一致。
圖1b所示为H-O径向分布函数与实验值。比较不同模型的峰值出现的位置,出现第一峰值和第二峰值的位置与实验值相吻合,第一峰值的出现是氢原子与氧原子形成的共价键产生的结果。
图1c所示为H-H径向分布函数与实验值。观察第一峰值的峰位和第二峰值的峰位,其分别反映的是中心水分子与近临水分子、第二配位圈水分子间的H-H距离。SPC和TIP3P模型的H-H径向分布函数与实验值吻合得不太理想,其他模型的模拟均与实验值吻合的较好。
综合分析径向分布函数,可以发现随着r的增加,函数趋于缓和,这种现象反映了水分子短程有序、长程无序的微观结构特征。综上所述,选用SPCE、TIP4P和TIP5P三种势能模型可以较好地反映出水分子的微观结构。
3.3 自扩散系数
可以用两种方法计算自扩散系数,一种是对均方位移(Mean Square Displacement,简称MSD)求斜率的Einstein方法[12],另一种是对速度自相关函数求积分的Green-Kubo方法。Einstein方法的公式为
其中,为水分子的质量,为玻尔兹曼常数,为体系的温度,、分别为0时刻和时刻粒子的速度。
采用以上两种方法分别计算水的自扩散系数。图2所示采用Einstein方法为不同模型计算的均方位移。可以看出均方位移斜率由小到大分别对应SPCE、TIP5P、TIP4P、SPC和TIP3P模型,表明自扩散系数也依次增大。
表3给出了采用两种方法计算的自扩散系数与其他文献[17]所得结果及实验值[18]的比较。可以看出,采用Einstein方法计算的自扩散系数要比Green-Kubo方法更为理想,所得结果比文献[17]的值更符合实验值。采用Einstein方法计算所得结果比Green-Kubo方法小,这与速度自相关函数的积分区间选取有关,从而导致了松弛时间的差异,即当速度自相关函数从1突降到0附近后,一直在0附近保持振荡,这表明此后粒子的速度与时间不再相关。松弛时间一般很难准确确定,这将会影响到自扩散系数积分区间的选取。SPCE模型得到的结果与实验值最接近,相对误差仅为0.39%。TIP5P模拟结果次之,相对误差为13.87%,但比SPC、TIP3P、TIP4P模拟结果要好得多。在TIP3P模型中,两种方法得到的结果与实验值相差最大,分析其原因可能是此模型下氢氧有效电荷数相差较大,使得氧原子束缚了氢原子。SPC和TIP4P计算结果也与实验值相差较大,但是相比文献[17]要好得多。
4 结束语
势能模型的选取是分子动力学计算的关键。本文采用分子动力学方法计算了五个势能模型下水分子的内能、汽化焓、径向分布函数及自扩散系数。
综合分析发现,SPCE模型最适用于水分子动力学性质的研究,能够较真实地反映水分子的热力学性质、微观结构和自扩散特性,值得学者广泛关注。
基金项目
教育后勤疫情防控专项课题(课题编号:ZXYBKT202022)
参考文献
[1] Alder B J, Wainwright T E. Phase Transition for a Hard Sphere System[J]. Journal of Chemical Physics, 1957, 27(5): 1208-1209.
[2] Rahman A. Correlations in the Motion of Atoms in Liquid Argon[J]. Physical Review, 1964, 136(2): 405-411.
[3] Cheung P S Y, Powles J G. The properties of liquid nitrogen. IV. A computer simulation[J]. Molecular Physics, 1975, 30: 921-949.
[4] Matsuoka O, Clementi E, Yoshimine M. CI study of the water dimer potential surface[J]. Journal of Chemical Physics, 1976, 64(4): 1351-1361.
[5] Stilliger F H, Rahman A. Improved simulation of liquid water by molecular dynamics[J]. Journal of Chemical Physics, 1974, 60(4): 1545-1557.
[6] Berendsen H J C, Postma J P M, Gunsteren W F, et al. Intermolecular Forces[M]. Holland: D. Reidel Publishing Company, 1981: 331.
[7] Berendsen H J C, Grigena J R, Straatsma T P. The missing term in effective pair potentials[J]. J Journal of Physical Chemistry, 1987, 91(24): 6269-6271.
[8] Mahoney M W, Jorgensen W L. A five-five model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions[J]. Journal of Chemical Physics, 2000, 112(20): 8910-8922.
[9] 李印实, 何雅玲, 孙杰, 等. 不同水分子模型凝结系数的分子动力学对比研究[J]. 西安交通大学学报, 2006, 40(11): 1272-1275.
[10] Mills R. Self-diffusion in normal and heavy water in the range 1-45.deg[J]. Journal of Physical Chemistry, 1973, 77(5): 685-688.
[11] Allen M P, Tildesley D J. Computer Simulation of liquids[M]. Oxford: Clareendon Press, 1987.
[12] De Leeuw S W, Perram J W, Smith E R. Simulation of electrostatic systems in periodic boundary conditions. I. Lattice sums and dielectric constant[C]// Proceedings of the Royal Society A, 1980, 373: 27-56.
[13] Gear C W. Numerical Integration of Ordinary Differential Equations[M]. New Jersey: Prentice Hill, 1971.
[14] Soper A K, Phillips M G. A new determination of the structure of water at 25℃[J]. Chemical Physics, 1986, 107(11): 47-60.
[15] 吴熊武, 腾藤, 李以圭, 等. 水—甲醇体系的Monte Carlo分子动力學模拟[J]. 化学学报, 1992, 50: 543-548.
[16] Krynicki K, Green C D, Sawyer D W. Pressure and temperature dependence of self-diffusion in water[J]. Faraday Discussions of the Chemical Society, 1978, 66: 199-208.
[17] Jorgensen W L, Jenson C. Temperature Dependence of TIP3P, SPC, and TIP4P Water from NPT Monte Carlo Simulations: Seeking Temperatures of Maximum Density[J]. Journal of Computational Chemistry, 1998, 19(10): 1179-1186.
[18] 刘娟芳, 曾丹苓, 蔡智勇, 等. 扩散系数的分子动力学模拟[M]. 工程热物理学报, 2006, 27(3): 373-375.
作者简介:
俞联梦(1983—),通信作者,女,云南大理人,硕士研究生,讲师,从事高校教学工作。主要研究方向:分子动力学。
E-mail: 175365186@qq.com
(收稿日期:2020-06-18)
猜你喜欢
科教新报(2021年11期)2021-05-12
科学之谜(2019年3期)2019-03-28
智富时代(2018年1期)2018-03-26
智富时代(2018年1期)2018-03-26
意林原创版(2017年11期)2017-12-01
中学生理科应试(2017年7期)2017-08-09
中学生理科应试(2017年7期)2017-08-09
中学物理·高中(2016年8期)2016-08-08
考试周刊(2016年1期)2016-03-11
中学生数理化·高一版(2008年4期)2008-11-15
