
植物长非编码RNA进化相关研究进展
2020-12-21 03:49桑世叶任强吴霜寒刘长宁
安徽农业科学 2020年22期
桑世叶 任强 吴霜寒 刘长宁
摘要 长非编码RNA(long non-coding RNA,lncRNA)的重要性在近年来的研究中日益凸显。由于lncRNA的同源性、保守性和特异性可以间接反映其生物学功能,越来越多的研究聚焦于理解lncRNA的进化过程。现主要就lncRNA基本性质、lncRNA鉴定方法、植物基因组进化以及植物lncRNA功能与进化的研究进展进行综述,以期为更好地开展植物lncRNA进化研究提供参考。
关键词 长非编码RNA;植物;基本性质;鉴定方法;功能;进化
中图分类号 Q943.2文献标识码 A文章编号 0517-6611(2020)22-0019-06
doi:10.3969/j.issn.0517-6611.2020.22.006
Research Progress on the Evolution of Long Non-coding RNA in Plants
SANG Shi-ye1,2,REN Qiang1,2, WU Shuang-han3 et al
(1.Xishuangbanna Tropical Botanical Garden,Chinese Academy of Sciences, Xishuangbanna, Yunnan 666303;2. University of Chinese Academy of Sciences, Beijing 100049;3.Sichuan Agricultural University, Chengdu, Sichuan 611130)
Abstract The importance of long non-coding RNA (lncRNA) has become increasingly prominent in recent years. Since the homology, conservation and specificity of lncRNA can indirectly reflect its biological functions, more and more researches focus on understanding the evolution process of lncRNA.This paper mainly reviewed the research progress on the basic properties of lncRNA, the identification methods of lncRNA, the evolution of plant genome and the function and evolution of plant lncRNA, in order to provide reference for the better development of the evolution of plant lncRNA.
Key words Long non-coding RNA;Plant;Basic properties;Identification methods;Function;Evolution
基金项目 国家自然科学基金面上项目(31970609)。
作者简介 桑世叶(1994—),女,四川成都人,硕士研究生,研究方向:植物长非编码RNA的进化分析。*通信作者,研究员,博士,博士生导师,从事长非编码RNA和生物信息学研究。
收稿日期 2020-04-03
转录组测序揭示了高等真核生物的绝大部分基因组序列是被转录的。然而大于75%的转录本不会被翻译为蛋白质,这些转录本被称为非编码RNAs(ncRNA)。长非编码RNAs(long non-coding RNAs,lncRNA)作为ncRNA的重要组成部分,被定义为长度大于200个核苷酸(nt)的ncRNA,獨立转录,但不具有编码功能蛋白的潜力。他们主要在RNA水平上发挥多种生物学功能,包括染色体剂量补偿效应、招募染色质重塑复合物到特定位点、模拟miRNAs靶标竞争性结合miRNAs、产生siRNA介导靶基因启动子区甲基化水平升高等。lncRNA在进化过程中具有保守性和特异性2个非常重要的生物学特征。lncRNA的保守性有3个维度,即序列保守性、结构保守性和位置保守性。lncRNA的特异性则包括种系特异性和组织特异性。由于lncRNA的同源性、保守性和特异性可以间接反映其生物学功能,越来越多的研究聚焦于理解lncRNA的进化过程。其中动物lncRNA的进化分析尤其受到关注,为理解动物lncRNA的进化历史、功能以及调控网络进化做出了重要贡献。反观植物领域,lncRNA在多物种中大规模进化分析的繁荣景象尚未出现,但仍有一些研究进行了相关探索。笔者通过对植物lncRNA的进化研究,以及与其息息相关的lncRNA生物学功能、lncRNA基因进化、植物基因组进化等多个研究领域进行阐述,以期为植物lncRNA的进化研究提供理论支撑和新的思考。
1 长非编码RNA 概述
1.1 长非编码RNA的特征和分类
遗传信息从基因序列到蛋白质的流动过程中,RNA承担着传递信息的重要角色。在测序技术尚未成熟时,人们普遍认为真核生物的基因组绝大部分是由编码蛋白的基因序列组成,而非编码区域则并不具有生物学功能。但事实证明,人们低估了非编码RNA(ncRNA)种类和功能的多样性。常见的非编码RNA 包括 tRNA、rRNA、microRNAs、piRNAs、snoRNAs、siRNAs、snRNAs、exRNAs、scaRNAs和lncRNAs等多种类型,其能通过在各个层次上调节从蛋白编码基因到蛋白质的遗传信息流,从而参与到生物体的各种生物学过程中。
lncRNA是一类长度大于200 nt,不具备编码功能蛋白潜力,主要由RNA 聚合酶Ⅱ(PolⅡ)转录生成,并具备甲基鸟苷帽子和多聚腺苷酸(poly-A)结构的ncRNA,其初级结构保守性差,二三级结构保守性很强,且在表达上具有极强的时空特异性。
由于lncRNA的研究还处于起步阶段,人们对它的认识还不够深入,科学界至今还没有统一的生物学分类标准。根据lncRNA与蛋白质编码基因的相对位置,可以将其分为:①反义型(antisense),此类lncRNA的反义链位置上是一个已知的蛋白编码基因;②内含子型(intronic),此类lncRNA处于蛋白编码基因的内含子区域;③正义型(divergent),此类lncRNA是由蛋白编码基因的正义链转录而来,序列内含蛋白编码基因的外显子;④基因间区型(intergenic),此类lncRNA处于蛋白编码基因之间,且与蛋白编码基因的距离在人类基因组上大于5 kb[1]。除此之外,还可以根据lncRNA的保守性,将其分为:①外显子-内含子结构保守型(conserved exonic structure),此类lncRNA的外显子-内含子结构和此结构处的多个序列在物种间保守;②序列保守型(conserved sequence),此类lncRNA是一类序列保守的lncRNA;③位置保守型(positionally conserved),此类lncRNA的上游或下游相似位置处具有直系同源的编码基因[2]。
1.2 长非编码RNA的生物学特性
在lncRNA发现的早期,人们对其生物学特性还缺乏充分的认识。由于蛋白编码基因的研究早已趋于成熟,所以研究者们尝试运用蛋白编码基因的研究手段来探索lncRNA的特性。但研究者们很快发现,lncRNA在序列结构、保守性、进化规律等方面都有很大的不同。近年来人们主要将对lncRNA生物学特性的研究重点放在保守性和特异性2个方面。
在lncRNA的保守性上,几乎所有的研究都得出了一致的结论,即lncRNA的保守性相较蛋白编码基因来说非常低。但研究lncRNA的保守性依旧为理解lncRNA做出了重要贡献,所以仍然具有重要的意义。lncRNA的保守性主要可以从序列保守性、结构保守性和位置保守性3个方面來研究[2]。lncRNA在这3个方面的保守性并非完全独立的,需要研究者们综合考虑并灵活选用。在序列保守性上,lncRNA序列保守性低仅是相较于蛋白编码基因序列而言的,如果与内含子序列和基因间区序列相比,lncRNA的序列仍具有较高的保守性。随着近年来转录组测序技术的发展,lncRNA的分析进入了超大规模的时代,对lncRNA的保守性低也出现了新的阐释。有研究指出,大多lncRNA的序列是具有保守性的,只是其保守性仅仅体现在一些小段的序列斑块上,且这些短序列大多出现在启动子区域,所以lncRNA的保守片段表现出5′端偏向性。在lncRNA行使功能时,只需要这些短序列发挥作用,所以表现出该lncRNA序列能够容忍基因结构重大变化的现象[3]。在结构保守性上,当仅使用序列保守来定义同源性而无法完全识别物种间的同源基因对时,通过结构保守就可以找到这些缺失的同源性的说法至今还没有证据可以证明。但是,利用全基因组比对的方法,可以预测序列中一些短的区域是在二级结构的约束下进化的,并且lncRNA外显子和这些短的区域的重叠部分在人类基因组和其他物种基因组中都很小。所以,虽然全基因组分析为寻找lncRNA中维持二级结构的选择压力仅提供了有限的支持,但这并不意味着基于结构的同源搜索对lncRNA同源检测没有用[2]。在位置保守性上,当比较远缘的物种时,有相当数量的lncRNA是位置保守的,即与同源蛋白编码基因和/或其他保守区域具有相同的相对方向,而它们中的大部分是不具有可检测的序列同源性[3-5]。这些位置保守的基因对可能对应于那些功能序列过短或因过短而无法检测到序列同源性的lncRNA,也可能对应于那些仅转录作用处于选择压力下的lncRNA。所以,位置保守性是对研究lncRNA保守性的有力补充。
在lncRNA的特异性上,其种系特异性和组织特异性则备受人们关注。在动物和植物中,lncRNA的种系特异性均有所发现。Derrien等[6]和Necsulea等[7]各自在对哺乳动物的lncRNA研究中均发现了大量灵长类特有的lncRNA。Washietl等[8]对6种哺乳动物中9个组织的lincRNA进行进化分析,发现一类人特有的富集于睾丸且进化速度非常快的lncRNA。Liu等[9]在拟南芥和其他植物物种间的比较分析表明,有98%的lncRNA是拟南芥特有的,只有少于2%的lncRNA是序列保守的。同时,还有很多研究证实了即使是同一组织中的lncRNA,它们在不同物种中的表达水平也会呈现出种系差异。由于lncRNA位点在进化过程中的获得和丢失是非常常见的,所以lncRNA的种系特异性往往被认为与物种的适应性进化有关。除了lncRNA的种系特异性外,其组织特异性的特征也尤为显著。Washietl等[8]研究发现在哺乳动物层面表达的lincRNAs表现出非常强的组织特异性。Hezroni等[3]通过对睾丸、脑、肝脏和肾4个组织的转录组数据进行比较分析,发现lncRNA在睾丸中的表达水平远远高于另外3个组织。Deng等[10]在植物中的研究也发现大量组织特异性表达的lncRNA,同时还发现不保守的lncRNA组织特异表达的比例高于保守的lncRNA。
1.3 长非编码RNA的生物学功能及调控方式
起初,lncRNA被认为是基因组转录的“噪音”,是RNA聚合酶Ⅱ的副产物,是不具有生物学功能的“垃圾”序列[11]。但越来越多的研究已经证实,lncRNA在生命调控网络中起着至关重要的作用,广泛参与DNA甲基化、组蛋白修饰、染色质重塑、细胞周期调控、mRNA降解、基因印记、增加mRNA的稳定性、调控丝氨酸、精氨酸剪接因子磷酸化等生物学过程,能与DNA、RNA、蛋白质分子作用,顺式或反式调控靶基因表达[12]。虽然人们已经逐渐意识到lncRNA在生命过程中发挥的巨大作用,但目前仍只有一小部分lncRNA被证实具有重大的生物学意义。例如,Xist 首先从X失活中心(X inactivation center,Xist)转录,通过招募PRC2靶向作用于X染色体特定位点,促进组蛋白H3第27位赖氨酸三甲基化(histone H3 lysine K27 trimethylation,H3K27me3),介导相关基因沉默[13-16]。而严重危害人类健康的癌症也被证实与lncRNA的调控密切相关,如印记基因H19,与正常组织相比在患癌组织中均呈现出高表达[17]。在水稻“农垦58S”中,一条名为LDMAR的lncRNA通过产生许多siRNAs使自身启动子甲基化水平升高从而无法转录,导致雄性光敏不育[18]。有研究表明,在植物的春化过程中,开花抑制基因FLC上转录的反义lncRNA(COLDAIR、COOLAIR)可以通过招募染色质重塑复合体PRC介导FLC染色质重塑来抑制FLC的表达,从而调控开花时间[19-20]。
尽管现有研究表明这些lncRNA可以通过直接或间接调控基因的表达来参与各种生物学过程,但绝大部分lncRNA的功能机制及其与生物体生长发育的关系尚不清楚。为了更好地理解lncRNA对基因表达的调控,有研究将lncRNA行使功能的作用方式分为以下几种:①在基因上游区域转录,从而干扰编码基因的转录;②介导染色质重塑和组蛋白修饰影响基因的表达;③调控可变剪切模式;④与Dicer酶共同作用产生内源siRNA调控基因的表达;⑤直接调节相关蛋白的活性影响蛋白发挥作用;⑥作为结构成分促使核酸蛋白复合体的形成;⑦改变蛋白质的定位;⑧与相关小分子如miRNA相互作用[21-22]。此后,李睿等[12]、王国峰[21]又将lncRNA调控基因表达分为3个层面:lncRNA参与基因表观遗传调控、lncRNA参与基因转录调控、lncRNA参与基因转录后调控,揭示了lncRNA可以在多个层面上以不同方式调控编码基因的表达,更加系统全面地概括了lncRNA参与生物学途径的方式。随着人们对lncRNA的了解更加深入,Wang等[23]在2011年首次提出将lncRNA行使生物学功能的方式分为4类:信号分子、诱饵分子、引导分子、支架分子,为推进lncRNA研究的发展做出了重大贡献。
1.4 长非编码RNA的起源
大量研究发现,无论是在动物还是植物基因组中的大多数lncRNA在经过千万年进化而分离的物种中没有同源物,表明lncRNA序列在物种间的保守性很低,这预示着新的lncRNA起源频率非常高。对于 lncRNA 的起源,Ulitsky[2]研究提出,物种进化过程中lncRNA的形成有5种机制:
①lncRNA序列复制产生新的lncRNA;
②蛋白质编码基因失去编码潜力产生新的lncRNA;
③经转座子整合后形成新的转录单元产生新的lncRNA;
④增强剪接信号的突变将具有转录潜力的序列转化为稳定的RNA,随后获得功能形成新的lncRNA;
⑤一系列适应成为新的lncRNA。
1.5 长非编码RNA的鉴定方法及鉴定流程
由于lncRNA的进化速率很快,序列保守性很低,且具有复杂多样的行使生物學功能的方式,所以很难寻求在基因组层面上鉴定lncRNA的算法。目前人们主要通过微阵芯片技术和转录组测序分析2种方法来鉴定lncRNA,由于微阵芯片技术存在交叉杂交和不能鉴定新的lncRNA等问题,所以仅仅用于早期的lncRNA鉴定。反之,随着近年来高通量测序技术和生物信息学的飞速发展,测序成本逐年降低,通过基因组和转录组测序来研究基因的进化和表达成为最主流的研究手段之一[24-25],相关的研究报道也是越来越多,使通过生物信息学方法分析RNA-seq数据,挖掘lncRNA序列、结构、进化、表达及功能等重要信息,并结合试验验证解析具有重要功能lncRNA调控机理的研究方法得到十分广泛的应用。
典型的RNA-seq工作流程的主要步骤包括总RNA提取、RNA富集、文库制备和测序。对于RNA富集,有2种方法,一是富集具有poly-A尾的RNA,二是去除核糖体RNA,保留剩余RNA。Poly-A RNA-Seq获得的RNA大约占一个物种总RNA的1/3,用于检测lncRNA的表达时是一种性价比较高的技术,但会遗漏掉不含有 poly-A 尾的 lncRNA。所以,目前使用较多的方法是去除核糖体RNA,此方法可以保留样本中完整的转录组,包括poly- A +和poly-A-转录本 。由于测序技术在实践过程中不能直接对RNA分子进行测序(目前已有技术可以做到,如纳米单分子测序技术),所以文库制备是进行测序之前的必要步骤,其目的主要有2个:①文库制备可以如实地代表样品中的RNA;②将RNA分子转化为更稳定的cDNA进行测序。
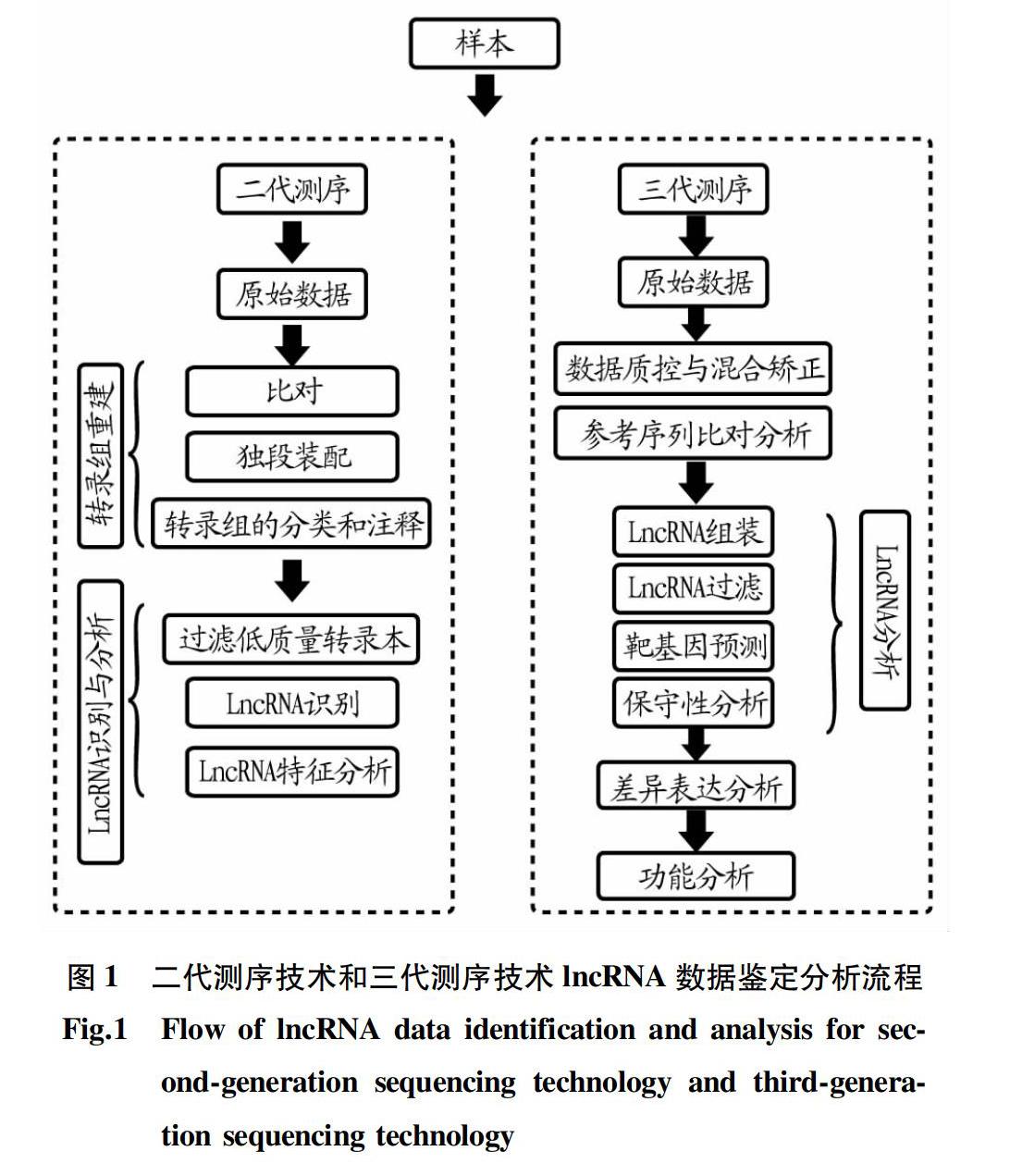
测序技术不断进步,第三代测序技术已经应运而生,也称下一代测序技术。第三代测序技术凭借着片段读长更长的优势在基因组研究中得到广泛应用,但由于测序通量低、测序成本贵、测序错误率高等缺点,它并没有完全占据市场,以Illumina平台为代表的第二代测序技术仍然普遍被应用。所以根据测序数据的来源,主要将lncRNA的鉴定分析流程大体上分为两类(图1):①通过Illumina测序技术产生的数据的鉴定分析流程;②依靠第三代测序技术进行的数据的鉴定分析。
鉴定分析流程中所涉及到的lncRNA的识别和预测,主要包含基本筛选和潜在编码能力筛选2个部分。基本筛选主要是根据lncRNA定义中对其长度大于200 nt的规定。潜在编码能力筛选所依据的原理主要是通过lncRNA的序列特征如碱基的排列、密码子的分布情况、组蛋白的修饰位点、序列的保守性等来预测该基因的蛋白编码潜能,从而区分蛋白编码基因和非蛋白编码基因。目前有多种lncRNA预测软件供研究者们使用,主要包括CPC2(coding potential calculator 2)、CNCI(coding-non-coding index)、PfamScan、CPAT(coding-potential assessment tool)等。 CPC2是一款非常流行的lncRNA预测软件,主要是利用Fickett TESTCODE分数、开放阅读框(ORF)长度、ORF完整性和等电点(pI)4个内在特征训练一个支持向量机(SVM)模型,从而预测转录本编码蛋白的潜力并对其进行分类。CPC2的运行速度比它的上一代CPC1快约1 000倍,并且与CPC1相比显示出更高的准确性,尤其是对于长非编码转录本而言[26]。此外,CPC2的模型是物种中性的,使其对于不断增长的非模式生物转录组是可行的。CNCI是中科院计算所赵屹团队开发的一款lncRNA预测工具,通过解析相邻的核苷酸三联体(adjoining nucleotide triplets,ANT),包括核苷酸三联体在编码域序列(CDS)和非编码RNA序列中的使用频率来构建SVM分类器,从而有效地区分蛋白质编码和非编码序列,而不依赖于已知的注释;这个工具适用于不完整的序列,如EST序列或从头拼接的转录本。Pfam是一个大型蛋白结构域家族的数据库,每个蛋白家族都由多个序列比对和隐马尔可夫模型(hidden Markovmodels,HMMs)所体现[27]。PfamScan可以通过分析基因的蛋白结构域来预测该基因的蛋白编码潜力,从而区分蛋白编码基因和非蛋白编码基因。CPAT使用了一个免比对的逻辑回归模型,该模型具有开放阅读框大小、开放阅读框覆盖率、Fickett TESTCODE统计量和六聚体使用偏差4个序列指标[28]。CPAT预测lncRNA具有优异的性能,准确度高,速度快。lncRNA的预测除了以上介绍的4种工具以外还有很多其他的预测工具,他们的核心思想都是通过估计基因的蛋白编码潜力来实现分类,只是具体的计算方式不一样。根据所持数据的不同情况,这些工具的预测准确度各有优劣,可以通过计算几种工具预测结果的交集作为最佳结果进行后续分析。
2 植物长非编码RNA进化与基因组进化的研究现状
2.1 长非编码RNA基因进化
现有动植物lncRNA进化的研究表明,与蛋白编码基因和miRNAs相比,lncRNA序列在物种间的保守性很低。例如,在小鼠和人类基因组中,大部分lncRNA在序列上的一致性都低于70%[29]。Liu等[9]研究发现,在拟南芥和其他植物物种间只有少于2%的lncRNA是序列保守的。这预示着新lncRNA的产生频率非常高,这种快速进化有助于组织和谱系特异性的 lncRNA 的出现,从而有利于物种之间形态差异的产生[30]。在真核生物中,生物体的复杂程度与基因组中 lncRNA含量的多少有关,而不是与整体 DNA 含量或编码基因的数目相关。因此,在基因组中lncRNA的扩增有利于复杂生物的进化[31]。由于 lncRNA 功能和分子进化的研究还处于初级阶段,lncRNA进化与物种特异特征之间的关系还有待进一步的研究。
2.2 植物长非编码RNA生物学功能
lncRNA 最先在人类中发现并报道,目前在人和动物中已经发现了很多lncRNA具有重要功能,如染色体剂量补偿效应、基因印记、器官形成、癌症等[32-33]。但在植物中仅少量lncRNA的功能被研究,大部分的研究都集中在对 lncRNA 的鉴定和功能预测上,包括其参与植物的生长发育过程、代谢过程和各种激素及胁迫的响应。最初,lncRNA的鉴定都集中在拟南芥、玉米、水稻等模式植物中,但随着测序成本的降低,目前有很多植物的lncRNA已经被鉴定,发展了很多关于植物lncRNA的数据库,典型的有GREENC、CANTATAdb、RefSeq等。
lncRNA开始对植物科学家们产生巨大吸引力很大程度上来自于人们对lncRNA调控植物开花过程的发现,这一过程是通过lncRNA调节春化作用来实现的,COOLAIR、COLDAIR和COLDWRAP就是这一过程中的典型代表。FLOWERING LOCUS C(FLC)基因是调控植物春化作用的关键基因,COOLAIR是FLC的反向转录本(NAT lncRNA),COLDAIR是来自于FLC的内含子区域的lincRNA,而COLDWRAP是转录于FLC的启动子上游的一条正义lncRNA。COOLAIR 的表达会招募相关蛋白清除FLC 上激活型组蛋白甲基标记,从而沉默FLC 转录本[34]。COLDAIR 则是通过结合PcG 蛋白复合体形成FLC染色质组蛋白抑制型甲基化,引起FLC 的沉默[35]。COLDWRAP可以参与调节春化作用介导的多梳复合物或影响FLC基因内染色质环的形成沉默或抑制FLC的表达[36]。3条lncRNA 都通过抑制FLC基因的表达参与调节春化作用,从而影响植物从营养生长向生殖生长的转变以及开花时间的早晚。除了上述3条与FLC有关的lncRNA外,拟南芥中还有一条名为FLORE的lncRNA也与开花过程相关,它可以通过抑制几种CDFs(CDF1、CDF3、CDF5)和增加FT转录水平来促进开花[36]。
生殖转变是植物生殖发育的第一步,lncRNA在生殖转变中具有如此重要的调控作用,暗示着lncRNA在植物生殖发育过程中的重要性。目前功能研究清楚的lncRNA中很多都有参与调控这一过程。例如,调控水稻长日照特异雄性不育的一个重要lncRNA LDMAR。LDMAR 会产生许多的siRNAs调控自身启动子甲基化水平升高从而无法转录,表达量的降低会造成未成熟花药的过早程序性细胞死亡,从而造成光敏型雄性不育[18]。拟南芥中的一条反义lncRNA asHSFB2a受热诱导会上调表达,从而调控拟南芥配子体发育过程中的热休克因子HSFB2a,最终影响配子体的发育[37]。除此之外,人们在其他物种中也发现有调节花粉发育的lncRNA,如在玉米中的一个雄蕊特异表达的lncRNA Zm401。Zm401基因的表达下调会显著影响花粉发育的关键基因ZmMADS2、MZm3-3和ZmC5的表达;导致小孢子和为花粉粒发育供应养分的绒毡层发育异常,最终导致玉米的雄性不育[38]。大白菜中也有一类名为BcMF11 的lncRNA 在调节花粉发育和雄蕊育性中行使功能,该lncRNA 长828 nt,在整个花粉发育阶段都有表达,当其表达降低时,绒毡层降解会延迟,造成花粉粒无法成熟[39-40]。
2.3 植物长非编码RNA进化
随着高通量测序技术的发展,越来越多的物种基因组被测序,使通过蛋白编码基因序列和表达图谱进化分析来研究家系特异表型遗传基础和单个基因功能成为可能。对于lncRNA,尽管转录组测序技术日渐成熟,但由于lncRNA没有易于计算的特征,注释不全,所以此类进化分析的研究仍然很匮乏,尤其是在植物中。近年来在人类、小鼠、拟南芥、水稻等生物中鉴定出了成千上万的lncRNA,尽管大部分lncRNA的功能还不清楚,但已经发现一部分lncRNA参与非常重要的生物学过程。所以发现哪些lncRNA有功能以及它们如何行使功能成为一个热门的科学问题,而lncRNA的进化研究是探索这一问题的重要科学手段。在基因的进化研究中,基因的同源性为比较不同物种在特定功能上或某个基因的遗传背景相似性提供了一个重要的研究线索。同源基因(homologous gene)是指由一个共同祖先在不同物种中遗传下来的基因,可以划分为直系同源基因和旁系同源基因。直系同源基因(orthologs)被定义为从同一祖先垂直进化而来的,作为物种形成的伴随事件而被重复,并通常继续保有相同生物学功能的基因[41]。旁系同源基因(paralogs)分为两类,一类是指由单个物种基因组中发生的各类复制事件而产生的基因,另一类旁系同源基因是指由于共同祖先的基因复制产生的并遗传到不同物种中的基因。旁系同源基因在功能上要么出现分化,要么产生计量补偿效应[41]。因为直系同源基因具有基因挖掘、基因注释、分子标记、协助构建基因间系统发生关系和追溯基因的进化历史等作用,所以目前已经被用于大量研究中,其鉴定方法也更加成熟。直系同源基因的識别方法主要可以总结为3类:①通过对比基因序列间的相似性来识别直系同源基因;②通过构建系统发育树推测直系同源关系;③结合基因序列比对和构建系统发育树2种方法来鉴定直系同源基因。
在动物中,Washietl等[8]分析发现在哺乳动物层面表达的lincRNAs表现出非常强的组织特异性的保守性,且相对于进化较年轻的lincRNA,在哺乳动物层面的lincRNA在启动子和外显子中表现出更高一级的序列保守性,还发现了一类人特有的富集于睾丸且进化速度非常快的lncRNA 。Necsulea等[7]对四足动物lncRNA进化的研究表明,尤其是古老的lncRNA,通常是被活跃调控的,可能主要在胚胎发育中起作用;lncRNA可能在精子发生、突触传递等基础过程和胎盘发育等具体机制中发挥功能。Hezroni等[3]研究发现,功能保守的lncRNA在序列上的保守性仅体现在一些小段的序列斑块的保守性上,该lncRNA行使功能只需要這些短序列发挥作用,以至于可以容忍基因结构的重大变化。Chen等[42]研究提出了一个可以鉴定高质量lncRNA并对其进行进化分析的工具slncky,进一步推进了lncRNA研究的发展。
在植物中,Liu等[9]研究发现在拟南芥中鉴定出的lncRNA一部分是器官特异性表达的,而其余的则是响应生物和非生物胁迫的,与其他6个植物物种相比,仅少于2%的lncRNA是进化保守的。Li等[43]通过对玉米和高粱lncRNA进行比较分析,发现只有25%的lncRNA是保守的。尽管大多数lncRNA的序列保守性很低,但对lncRNA进行全局性统计分析的研究表明,lncRNA在共线性和基因结构上具有进化保守性[4,10,24]。例如,Nitsche等[24]研究通过剪接位点的保守性来追踪lncRNA的进化,发现超过85%的人类lncRNA出现在胎盘哺乳动物的分化中。Deng等[10]对10种植物保守性分析的研究发现,序列和位置不保守的lncRNA组织特异表达的比例高于保守的lncRNA,表明保守的lncRNA比不保守的lncRNA更倾向于构成表达。
2.4 植物基因组进化
与其他真核生物基因组不同,植物基因组往往以更高的速率进化,从而导致更高的基因组多样性[44-45]。例如,亲缘关系密切的植物物种之间基因组大小的差异要比亲缘关系密切的动物物种之间的差异大得多[46]。同时,与动物不同,古老的复制事件、复制基因的高保存率以及全基因组加倍事件导致了植物基因组中存在大量的复制基因[46]。
基因复制是基因组序列和蛋白-蛋白相互作用(PPI)网络生长的重大进化事件。它被认为是形成和重构生物体功能的主要贡献者,因此得到了广泛的研究,特别是就其在进化中的作用而言[47]。基因复制最初具有冗余功能,可能改变基因剂量和/或重塑基因组结构,对基因的进化起到了至关重要的作用[48]。lncRNA基因的进化作为基因组进化的一部分,复制事件也必然与其有着密不可分的联系。
就对基因组的影响程度而言,全基因组复制是最具颠覆性的基因复制形式,它涉及到整个染色体或整个基因组的复制,对现存的复制基因贡献最大[46],因此被认为是物种多样性的主要驱动力[48]。在动物中,最近的全基因组加倍事件发生在大约450百万年前的人类谱系和大约200百万年前的出芽酵母谱系中[49-50]。而在被子植物中,全基因组加倍事件在过去200百万年的进化过程中发生了很多次[51-55],并且基因组测序还在继续揭露更多的全基因组加倍事件[56-61],这可以合理地解释多倍体和旁系同源基因在植物物种中的普遍存在。例如,全基因组复制导致芜菁[62]、野萝卜[63]、小麦[64]的三倍体化以及栽培草莓[65]的八倍体化。一项鉴定41个陆地植物基因组序列中重复基因的研究表明,平均64.5%的植物基因是旁系同源的,从苔藓植物小立碗藓的45.5%到苹果的84.4%[46]。
从以上研究可以看出,相对于动物来说,植物基因组的进化要快得多,这可以合理地解释植物lncRNA保守性较差、起源时间较晚的现象。所以,植物基因组的进化对研究植物lncRNA的进化保守性是具有很大的启发和指导意义的。
3 总结
越来越多的研究表明,lncRNA可以以多种调控方式调节基因的表达,参与各种各样的生物学过程,无论是在基础理论还是在实际应用中,都足以证明lncRNA的重要性和对其进行研究的必要性。目前对lncRNA的研究大部分都集中在某些物种的某些lncRNA的功能研究或者lncRNA的序列结构和鉴定上,对lncRNA进化的研究很少,且注意力大多都放在了动物中。所以植物lncRNA的进化研究仍是一个重要、新颖且急待人们去探索的科学问题。目前高通量测序技术飞速发展,越来越多的植物物种被测序,有了完整的基因组数据。同时,基于表达序列标签(expressed sequence tag)和全长cDNA测序(full length cDNA sequencing),微阵列芯片技术(tiling microarrays)和转录组测序(RNA-seq)的基因组学研究也已经逐步完善,为研究lncRNA的进化研究提供了良好的数据基础。因此,采用生物信息学的方法大规模分析植物lncRNA数据,探寻植物lncRNA的进化规律成为一个重要的研究方向。此问题上的突破,必将推动植物全基因组水平上进化系统的研究,发掘和研究出更多植物lncRNA的功能,从而对lncRNA产生更加深刻的认识。
参考文献
[1] MA L N,BAJIC V B,ZHANG Z.On the classification of long non-coding RNAs[J].RNA Biol,2013,10(6):925-933.
[2] ULITSKY I.Evolution to the rescue:Using comparative genomics to understand long non-coding RNAs[J].Nat Rev Genet,2016,17(10):601-614.
[3] HEZRONI H,KOPPSTEIN D,SCHWARTZ M,et al.Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species[J].Cell Rep,2015,11(7):1110-1122.
[4] MOHAMMADIN S,EDGER P P,PIRES J C,et al.Positionally-conserved but sequence-diverged:Identification of long non-coding RNAs in the Brassicaceae and Cleomaceae[J].BMC Plant Biol,2015,15:1-12.
[5] ULITSKY I,SHKUMATAVA A,JAN C H,et al.Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution[J].Cell,2011,147(7):1537-1550.
[6] DERRIEN T,JOHNSON R,BUSSOTTI G,et al.The GENCODE v7 catalog of human long noncoding RNAs:Analysis of their gene structure,evolution,and expression[J].Genome Res,2012,22(9):1775-1789.
[7] NECSULEA A,SOUMILLON M,WARNEFORS M,et al.The evolution of lncRNA repertoires and expression patterns in tetrapods[J].Nature,2014,505(7485):635-640.
[8] WASHIETL S,KELLIS M,GARBER M.Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals[J].Genome Res,2014,24(4):616-628.
[9] LIU J,JUNG C,XU J,et al.Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis[J].Plant Cell,2012,24(11):4333-4345.
[10] DENG P C,LIU S,NIE X J,et al.Conservation analysis of long non-coding RNAs in plants[J].Sci China Life Sci,2018,61(2):190-198.
[11] PONTING C P,OLIVER P L,REIK W.Evolution and functions of long noncoding RNAs[J].Cell,2009,136(4):629-641.
[12] 李睿,楊永芳,李冉,等.长链非编码RNA的功能及其作用机制[J].生命科学,2016,28(6):703-711.
[13] MAENNER S,BLAUD M,FOUILLEN L,et al.2-D structure of the A region of Xist RNA and its implication for PRC2 association[J].PLoS Biol,2010,8(1):1-16.
[14] ARTHOLD S,KUROWSKI A,WUTZ A.Mechanistic insights into chromosome-wide silencing in X inactivation[J].Hum Genet,2011,130(2):295-305.
[15] ESCAMILLA-DEL-ARENAL M,DA ROCHA S T,HEARD E.Evolutionary diversity and developmental regulation of X-chromosome inactivation[J].Hum Genet,2011,130(2):307-327.
[16] JEON Y,LEE J T.YY1 tethers Xist RNA to the inactive X nucleation center[J].Cell,2011,146(1):119-133.
[17] GIBB E A,BROWN C J,LAM W L.The functional role of long non-coding RNA in human carcinomas[J].Molecular cancer,2011,10(1):1-17.
[18] DING J H,LU Q,OUYANG Y D,et al.A long noncoding RNA regulates photoperiod-sensitive male sterility,an essential component of hybrid rice[J].Proc Natl Acad Sci USA,2012,109(7):2654-2659.
[19] KIM D H,XI Y P,SUNG S.Modular function of long noncoding RNA,COLDAIR,in the vernalization response[J].PLoS Genet,2017,13(7):1-18.
[20] CSORBA T,QUESTA J I,SUN Q W,et al.Antisense COOLAIR mediates the coordinated switching of chromatin states at FLC during vernalization[J].Proc Natl Acad Sci USA,2014,111(45):16160-16165.
[21] 王國峰.禾本科植物lncRNA的鉴定与分析[D].福州:福建农林大学,2018:2.
[22] LIN T,ZHU G T,ZHANG J H,et al.Genomic analyses provide insights into the history of tomato breeding[J].Nature genetics,2014,46(11):1220-1226.
[23] WANG K C,CHANG H Y.Molecular mechanisms of long noncoding RNAs[J].Mol Cell,2011,43(6):904-914.
[24] NITSCHE A,ROSE D,FASOLD M,et al.Comparison of splice sites reveals that long noncoding RNAs are evolutionarily well conserved[J].RNA,2015,21(5):801-812.
[25] LIN T,ZHU G T,ZHANG J H,et al.Genomic analyses provide insights into the history of tomato breeding[J].Nat Genet,2014,46(11):1220-1226.
[26] KANG Y J,YANG D C,KONG L,et al.CPC2:A fast and accurate coding potential calculator based on sequence intrinsic features[J].Nucleic Acids Res,2017,45:W12-W16.
[27] EL-GEBALI S,MISTRY J,BATEMAN A,et al.The Pfam protein families database in 2019[J].Nucleic Acids Res,2019,47:D427-D432.
[28] WANG L G,PARK H J,DASARI S,et al.CPAT:Coding-potential assessment tool using an alignment-free logistic regression model[J].Nucleic Acids Res,2013,41(6):1-7.
[29] PANG K C,FRITH M C,MATTICK J S.Rapid evolution of noncoding RNAs:Lack of conservation does not mean lack of function[J].Trends Genet,2006,22(1):1-5.
[30] KUTTER C,WATT S,STEFFLOVA K,et al.Rapid turnover of long noncoding RNAs and the evolution of gene expression[J].PLoS Genet,2012,8(7):1-15.
[31] SUN X M,TANG Y P,MENG X Z,et al.Sequencing and analysis of a genomic fragment provide an insight into the Dunaliella viridis genomic sequence[J].Acta Biochim Biophys Sin,2006,38(11):812-820.
[32] AZEVEDO-MARTINS A C,MACHADO A C L,KLEIN C C,et al.Mitochondrial respiration and genomic analysis provide insight into the influence of the symbiotic bacterium on host trypanosomatid oxygen consumption[J].Parasitology,2015,142(2):352-362.
[33] EWART K M,JOHNSON R N,OGDEN R,et al.Museum specimens provide reliable SNP data for population genomic analysis of a widely distributed but threatened cockatoo species[J].Mol Ecol Resour,2019,19(6):1578-1592.
[34] HAN S Y,LIANG Y C,LI Y,et al.Lncident:A tool for rapid identification of long noncoding RNAs utilizing sequence intrinsic composition and open reading frame information[J].Int J Genomics,2016,2016:1-11.
[50] DEHAL P,BOORE J L.Two rounds of whole genome duplication in the ancestral vertebrate[J].PLoS Biol,2005,3(10):1700-1708.
[51] LYONS E,PEDERSEN B,KANE J,et al.Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya,poplar,and grape:CoGe with rosids[J].Plant Physiol,2008,148(4):1772-1781.
[52] SOLTIS D E,ALBERT V A,LEEBENS-MACK J,et al.Polyploidy and angiosperm diversification[J].Am J Bot,2009,96(1):336-348.
[53] LEE T H,TANG H B,WANG X Y,et al.PGDD:A database of gene and genome duplication in plants[J].Nucleic Acids Res,2013,41:1152-1158.
[54] RENNY-BYFIELD S,WENDEL J F.Doubling down on genomes:Polyploidy and crop plants[J].Am J Bot,2014,101(10):1711-1725.
[55] SOLTIS D E,VISGER C J,SOLTIS P S.The polyploidy revolution then...and now:Stebbins revisited[J].Am J Bot,2014,101(7):1057-1078.
[56] VELASCO R,ZHARKIKH A,AFFOURTIT J,et al.The genome of the domesticated apple(Malus x domestica Borkh.)[J].Nat Genet,2010,42(10):833-839.
[57] DHONT A,DENOEUD F,AURY J M,et al.The banana(Musa acuminata)genome and the evolution of monocotyledonous plants[J].Nature,2012,488(7410):213-217.
[58] WANG Z W,HOBSON N,GALINDO L,et al.The genome of flax(Linum usitatissimum)assembled de novo from short shotgun sequence reads[J].Plant J,2012,72(3):461-473.
[59] LU F,LIPKA A E,GLAUBITZ J,et al.Switchgrass genomic diversity,ploidy,and evolution:Novel insights from a network-based SNP discovery protocol[J].PLoS Genet,2013,9(1):1-14.
[60] MYBURG A A,GRATTAPAGLIA D,TUSKAN G A,et al.The genome of Eucalyptus grandis[J].Nature,2014,510(7505):356-362.
[61] WANG W,HABERER G,GUNDLACH H,et al.The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle[J].Nat Commun,2014,5:1-13.
[62] LYSAK M A,KOCH M A,PECINKA A,et al.Chromosome triplication found across the tribe Brassiceae[J].Genome Res,2005,15(4):516-525.
[63] MOGHE G D,SHIU S H.The causes and molecular consequences of polyploidy in flowering plants[J].Ann N Y Acad Sci,2014,1320(1):16-34.
[64] SALSE J,BOLOT S,THROUDE M,et al.Identification and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution[J].Plant Cell,2008,20(1):11-24.
[65] BYRNE D H,JELENKOVIC G.Cytological diploidization in the cultivated octoploid strawberry Fragaria × ananassa[J].Can J Genet Cytol,1976,18(4):653-659.
猜你喜欢
中国科技论坛(2017年7期)2017-07-25
爆笑show(2016年7期)2017-02-09
少儿科学周刊·儿童版(2015年10期)2015-11-07
少儿科学周刊·儿童版(2015年1期)2015-07-07
中国中医药现代远程教育(2014年22期)2014-03-01
中国中医药现代远程教育(2014年16期)2014-03-01
