
水蒸气蒸馏-气相色谱-质谱法测定皮革制品中19种含氯苯酚
2020-12-12 08:17:20,,,
理化检验-化学分册 2020年11期
,,,
(黎明职业大学材料与化学工程学院,泉州 362000)
含氯苯酚是一类具有持久高毒性的苯酚类化合物,根据含氯苯酚中氯原子取代氢原子的数目及位置的不同,可将其分为一氯苯酚(MoCP)、二氯苯酚(DiCP)及其同分异构体、三氯苯酚(TriCP)及其同分异构体、四氯苯酚(TeCP)及其同分异构体、五氯苯酚(PCP),共19种。含氯苯酚常用作皮革及其制品的防腐、防霉、防蛀剂[1]。穿着含有过量含氯苯酚的服装和鞋子时,含氯苯酚可通过皮肤在人体内蓄积,具有潜在的致癌、致畸风险,不仅对人类的健康产生威胁,还会造成生态环境的污染和破坏[2]。许多发达国家和国际权威组织相继颁布技术法规和标准,对含氯苯酚的含量加以控制。Oeko-Tex Standard 100(2016)和GB/T 18885-2009对含氯苯酚的限量为婴儿类用品不大于0.05 mg·kg-1,其他类用品不大于0.5 mg·kg-1。
皮革制品中的含氯苯酚一般以含氯苯酚及其盐类的形式存在[ISO 17070-2015(E)],一般通过有机溶剂或碱性溶液超声法进行萃取。超声萃取法具有操作简单,适合批量处理样品的优点,也有萃取液基质复杂,萃取效率不完全等缺点。文献[3]发现,采用丙酮萃取时易将皮革上的染料组分萃取下来,对后续仪器分析造成一定的难度。文献[4]尝试用氢氧化钾溶液超声萃取时发现,氧氧化钾溶液对皮革样品中有机物的浸润和溶解作用较差,萃取效率较低。水蒸气蒸馏法用水蒸气提取不溶或难溶于水但有一定挥发性的有机物,使该物质在低于100 ℃时随着水蒸气一起蒸馏出来,是一种分离和提纯有机物的重要方法。常用的水蒸气蒸馏装置包括水蒸气发生器、蒸馏烧瓶、冷凝管、接收器等4个部分。
皮革制品中含氯苯酚的测定方法主要有气相色谱法[5]、气相色谱-质谱联用法[6]、液相色谱法[7]、液相色谱-串联质谱法[8]。采用气相色谱法时,需将含氯苯酚转化为含氯苯酚乙酰化产物,再进行气相色谱分析,它使用的电子捕获检测器灵敏度高,但其定性分析能力不如气相色谱-质谱法的,且低氯目标物(MoCP、DiCP)的响应太低。液相色谱法虽然不需要衍生化,但易产生假阳性结果。液相色谱-串联质谱法仪器价格昂贵,普及率较低。
目前,相关文献的分析对象多为纺织品,且仅对其中的TriCP、TeCP、PCP进行研究[9-10]。本工作采用水蒸气蒸馏-气相色谱-质谱法测定皮革制品中的19种含氯苯酚含量,以期为皮革中含氯苯酚的同步提取和测定提供技术支撑。
1 试验部分
1.1 仪器与试剂
Agilent 7890A-5975C型气相色谱-质谱仪;MMV-1000W型分液漏斗振荡器;BSA 223S型电子天平;98-1-B型电子调温电热套;Milli-Q型超纯水系统;带聚四氟乙烯隔垫瓶盖的管状硬质玻璃提取瓶(60mL)。
19种含氯苯酚混合标准溶液:2-氯苯酚(2-MoCP)、3-氯苯酚(3-MoCP)、4-氯苯酚(4-MoCP)、2,6-二氯苯酚(2,6-DiCP)、2,5-二氯苯酚(2,5-DiCP)、2,4-二氯苯酚(2,4-DiCP)、3,5-二氯苯酚(3,5-DiCP)、2,3-二氯苯酚(2,3-DiCP)、3,4-二氯苯酚(3,4-DiCP)、2,4,6-三氯苯酚(2,4,6-TriCP)、2,3,6-三氯苯酚(2,3,6-TriCP)、2,3,5-三氯苯酚(2,3,5-TriCP)、2,4,5-三氯苯酚(2,4,5-TriCP)、2,3,4-三氯苯酚(2,3,4-TriCP)、3,4,5-三氯苯酚(3,4,5-TriCP)、2,3,5,6-四氯苯酚(2,3,5,6-TeCP)、2,3,4,6-四氯苯酚(2,3,4,6-TeCP)、2,3,4,5-四氯苯酚(2,3,4,5-TeCP)、五氯苯酚(PCP)的质量浓度均为50 mg·L-1,介质为丙酮,使用时用丙酮逐级稀释至所需质量浓度。
四氯邻甲氧基苯酚(TCG)内标溶液:2 mg·L-1,介质为丙酮。
硫酸溶液:1 mol·L-1。
碳酸钾溶液:0.1 mol·L-1。
混合标准溶液系列:取5只60mL管状硬质玻璃提取瓶,加入0.1 mol·L-1碳酸钾溶液25mL,2 mg·L-1的TCG内标溶液0.125mL,再分别加入1 mg·L-1的19种含氯苯酚混合标准溶液0.005,0.025,0.05,0.1,0.2mL,混匀后依次加入乙酸酐0.3mL、三乙胺0.1mL、正己烷5mL,以320 r·min-1的转速在分液漏斗振荡器上振荡30 min,静置分层,取上层有机相约1mL到进样瓶中,得到1,5,10,20,40μg·L-1的混合标准溶液系列。
丙酮、正己烷为色谱纯;无水碳酸钾、乙酸酐、三乙胺为分析纯;硫酸为优级纯;试验用水为超纯水。
1.2 仪器工作条件
1)色谱条件 DB-5MS超高惰性毛细管色谱柱(20 m×0.18 mm,0.18μm);进样口温度280℃;脉冲不分流进样,脉冲压力200 k Pa,脉冲时间0.75 min;进样量1μL;载气为氦气,纯度不小于99.999%;流量0.72mL·min-1;溶剂延迟时间3.20 min。柱升温程序:初始温度80 ℃,保持0.6 min;以25 ℃·min-1速率升温至250 ℃,保持0.5 min。
2)质谱条件 电子轰击(EI)离子源,电离能量70 e V;传输线温度280℃,离子源温度230℃,四极杆温度150 ℃;全扫描范围质荷比(m/z)40~400;选择离子监测(SIM)模式。
目标物和内标物乙酰化产物的保留时间及其他质谱参数见表1。
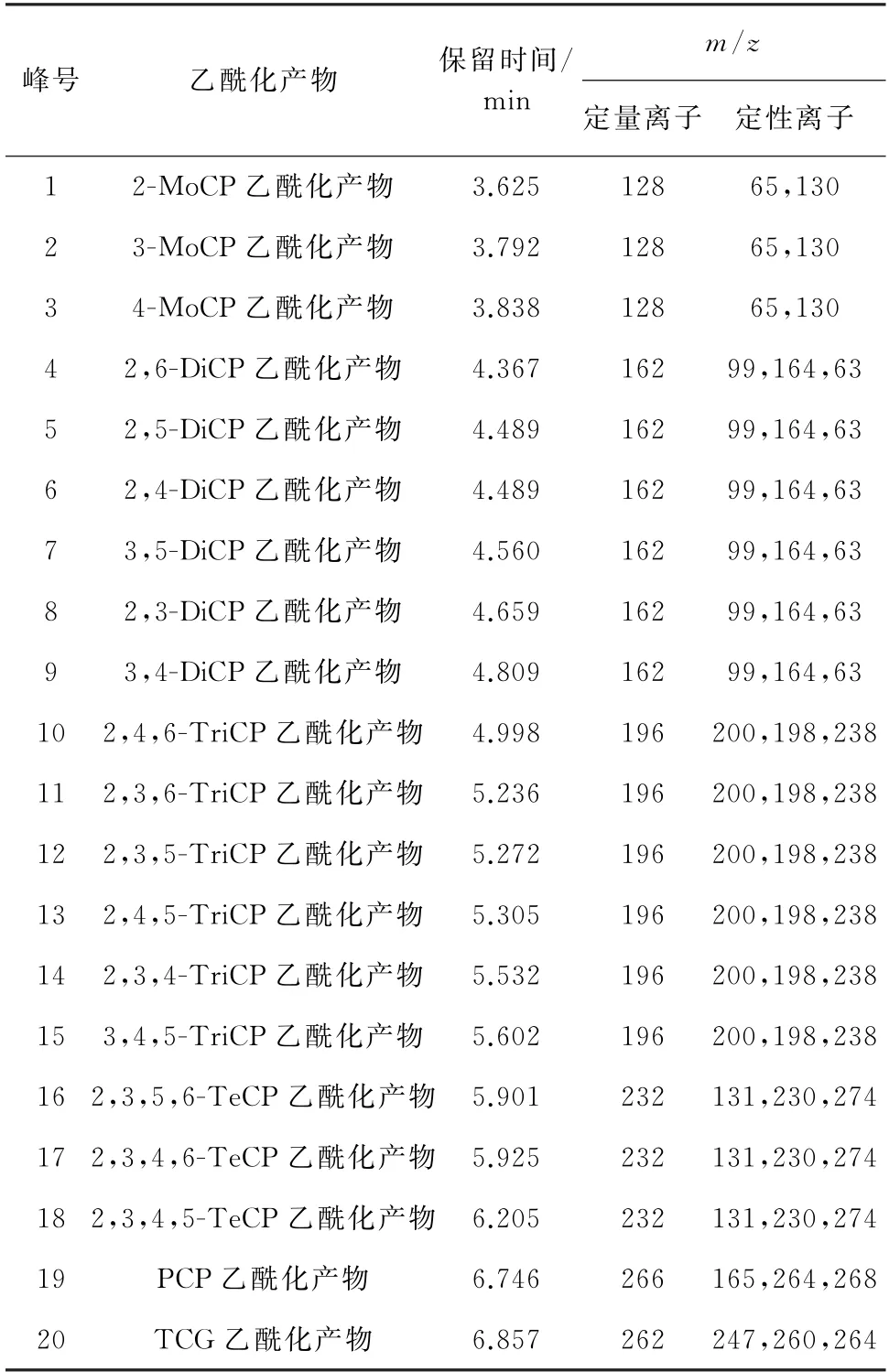
表1 保留时间及其他质谱参数Tab.1 Retention times and other MS parameters
1.3 试验方法
将皮革样品剪成5 mm×5 mm以下的小块,称取1.000 g样品于250mL圆底烧瓶中,加入硫酸溶液20mL,再加入TCG内标溶液1mL,置于铁架台上。在1 L圆底蒸馏烧瓶中加水至2/3处,作为水蒸气发生器,置于电热套上,通过玻璃导管与样品瓶相连。同时将样品瓶通过玻璃导管与冷凝管相连,用装有5 g无水碳酸钾的500mL烧杯作为接收器,收集蒸出的皮革样品中的含氯苯酚与水蒸气。
当收集的馏出液接近450mL后,停止加热,将馏出液转移至500mL容量瓶中,用水定容至刻度,摇匀静置。分取25mL于60mL管状硬质玻璃提取瓶中,依次加入乙酸酐0.3mL、三乙胺0.1mL、正己烷2mL,以320 r·min-1的转速在分液漏斗振荡器上振荡30 min,静置分层,取上层有机相约1mL到进样瓶中,按照仪器工作条件测定。
2 结果与讨论
2.1 色谱柱的选择
为了实现19种含氯苯酚中同分异构体的有效分离,需要选择合适的色谱柱及柱升温程序。试验考察超高惰性 DB-5MS(20 m ×0.18 mm,0.18μm)、超高惰性DB-35MS(20 m×0.18 mm,0.18μm)和超高惰性 DB-EUPAH (20 m ×0.18 mm,0.14μm)等3种色谱柱对19种含氯苯酚乙酰化产物分离情况的影响。结果发现,3种色谱柱对MoCP、TriCP、TeCP的同分异构体的乙酰化产物和PCP的乙酰化产物均有很好的分离效果,但对DiCP同分异构体的乙酰化产物的分离效果各有差异。在中等极性色谱柱DB-35MS上,2,6-DiCP和3,5-DiCP的乙酰化产物的保留时间重叠,2,5-DiCP和2,4-DiCP的乙酰化产物的保留时间也重叠;在中等极性色谱柱DB-EUPAH上,2,6-DiCP、2,5-DiCP和2,4-DiCP的乙酰化产物的保留时间重叠,而且这一现象在对柱升温程序进行优化后也难以解决,可能是由这4种同分异构体间的化学性质差别较小所致。在弱极性色谱柱DB-5MS(20 m×0.18 mm,0.18μm)上,除了2,5-DiCP和2,4-DiCP乙酰化产物无法分离外,其他17种含氯苯酚乙酰化产物均能完全分离,可能的原因是含氯苯酚乙酰化产物为弱极性物质,在弱极性色谱柱上有更好的分离效果。因此,试验选用DB-5MS毛细管色谱柱(20 m×0.18 mm,0.18μm)。
2.2 质谱条件的选择
对40μg·L-1混合标准溶液进行全扫描模式分析,提取质谱图,根据其特征离子碎片,选择响应较高的特征离子作为定性和定量离子,同时确定保留时间。为了减少基质干扰,提高检测方法的选择性和灵敏度,试验选用SIM模式进行分析。在优化的试验条件下,含氯苯酚及内标物乙酰化产物的SIM色谱图见图1。
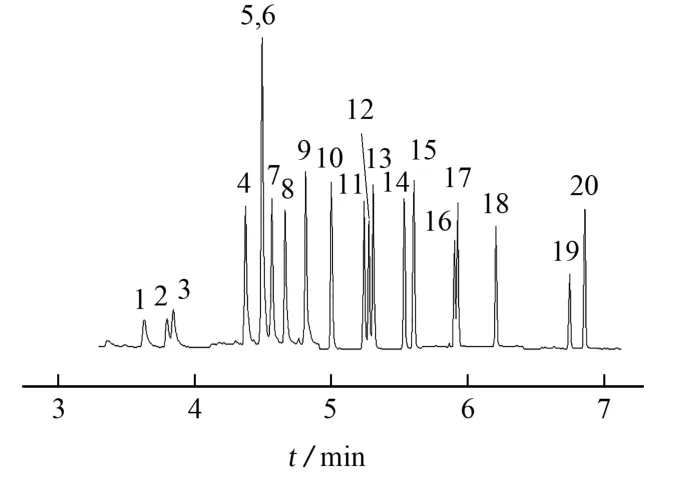
图1 含氯苯酚及内标物乙酰化产物的SIM色谱图Fig.1 SIM chromatogram of acetylation products of chlorinated phenol and internal standard
2.3 萃取条件的选择
皮革中的含氯苯酚可通过水蒸气蒸馏法直接萃取,但含氯苯酚盐易溶于水,在水溶液中呈电离态,蒸馏提取困难。在p H小于3的强酸溶液中,含氯苯酚盐会与强酸反应生成弱酸性难溶于水的含氯苯酚[11],可实现水蒸气蒸馏法的萃取。试验分别考察了采用硝酸、盐酸、硫酸为强酸性介质时,对蒸馏萃取效率的影响,试验发现:硝酸受热易挥发和分解,容易被蒸馏出来,影响蒸馏萃取效果;盐酸也具有挥发性,无法持续提供强酸环境;硫酸为高沸点的强氧化性酸,可持续提供强酸环境,将含氯苯酚盐转化为含氯苯酚随水蒸气蒸出,并且可氧化和腐蚀皮革制品表面,有利于水蒸气渗透,提高萃取效率。因此,试验选择1 mol·L-1硫酸溶液作水蒸气蒸馏试验的强酸性介质。
试验还考察了馏出液体积分别为150,250,350,450mL时对含氯苯酚乙酰化产物峰面积的影响,结果见图2。
图2结果表明:馏出液体积对峰面积有显著影响,馏出液体积越大,萃取效率也越高。在馏出液体积为450mL时,其中4-MoCP、3,4-DiCP的峰面积呈微弱的上升趋势,另外3种目标物的峰面积呈下降趋势,但变化幅度均不大。综合考虑,试验选择馏出液体积为450mL。
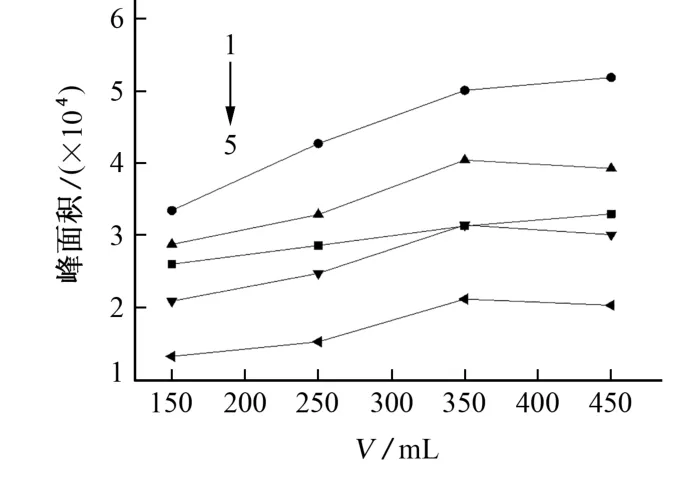
图2 馏出液体积对含氯苯酚乙酰化产物的峰面积的影响Fig.2 Effect of distillate volume on peak area of acetylation products of chlorinated phenol
由于含氯苯酚难溶于水,在馏出液中不能形成稳定且均匀的溶液,而无水碳酸钾可与含氯苯酚反应生成含氯苯酚钾盐,可稳定存在于馏出液中,保证了测试溶液的代表性和测定结果的准确度。
对含氯苯酚阳性样品进行水蒸气蒸馏试验,考察了蒸馏次数分别为1次和2次时对测定结果的影响。结果表明:第1次蒸馏得到的含氯苯酚目标物为2,4,6-TriCP、2,3,4,6-TeCP和PCP,质量分数分别为0.089,0.459,0.354 mg·kg-1;第2次蒸馏样品残渣得到的含氯苯酚目标物的测定值低于检出限,表明该方法经1次蒸馏即可基本提取完全。
2.4 衍生化条件的选择
含氯苯酚含有羟基,分子的极性较强,不利于色谱分离,因此,在前处理中,需将含氯苯酚衍生化后再进行仪器分析[12]。含氯苯酚衍生化常用的方法为乙酸酐乙酰化,该方法操作简单,衍生物的稳定性相对较好。衍生后的分子极性大大降低,拖尾峰消除,峰形改善。
为保证定量结果的准确和乙酰化反应完全,乙酸酐加入量必须过量,试验选择乙酸酐加入量为0.3mL,远大于皮革样品中可能存在的含氯苯酚乙酰化所需要的量。
乙酸酐乙酰化反应速度较慢,因此试验考察了衍生时间分别为10,20,30,40 min时对25μg·L-1混合标准溶液衍生效果的影响,结果见图3。
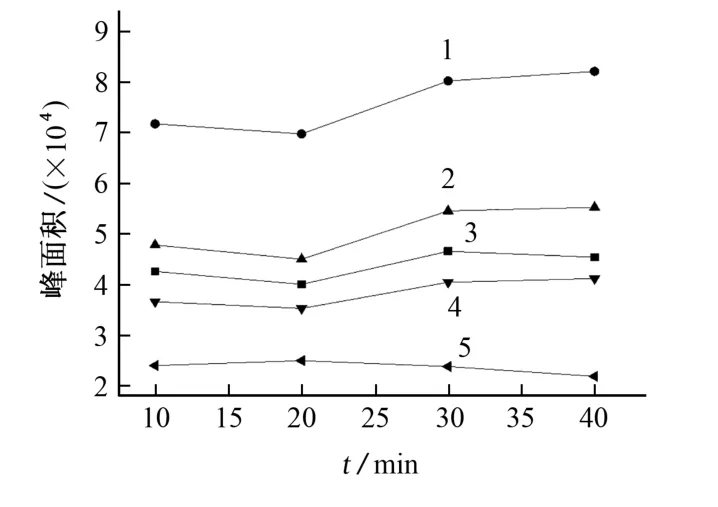
图3 衍生时间对含氯苯酚乙酰化产物的峰面积的影响Fig.3 Effect of derivative time on peak area of acetylation products of chlorinated phenol
结果表明:衍生后得的含氯苯酚乙酰化产物的峰面积随衍生时间的延长而增大,当衍生时间超过30 min后,峰面积变化不大,因此试验选择衍生时间为30 min。
2.5 标准曲线和检出限
按照仪器工作条件对混合标准溶液系列进行测定,以含氯苯酚乙酰化产物与内标物乙酰化产物质量浓度之比为横坐标,两者的定量离子的峰面积之比为纵坐标绘制标准曲线,当含氯苯酚的质量浓度为1~40μg·L-1时,其对应的乙酰化产物和内标物乙酰化产物的质量浓度之比和相应的峰面积比值呈线性关系,其他线性参数见表2。
按照试验方法对11个加标量为0.05 mg·kg-1的空白皮革样品进行测定,以3倍标准偏差(s)计算检出限(3s),10倍标准偏差计算测定下限(10s),所得结果见表2。

表2 线性参数、检出限和测定下限Tab.2 Linearity parameters,detection limits and lower limits of determination
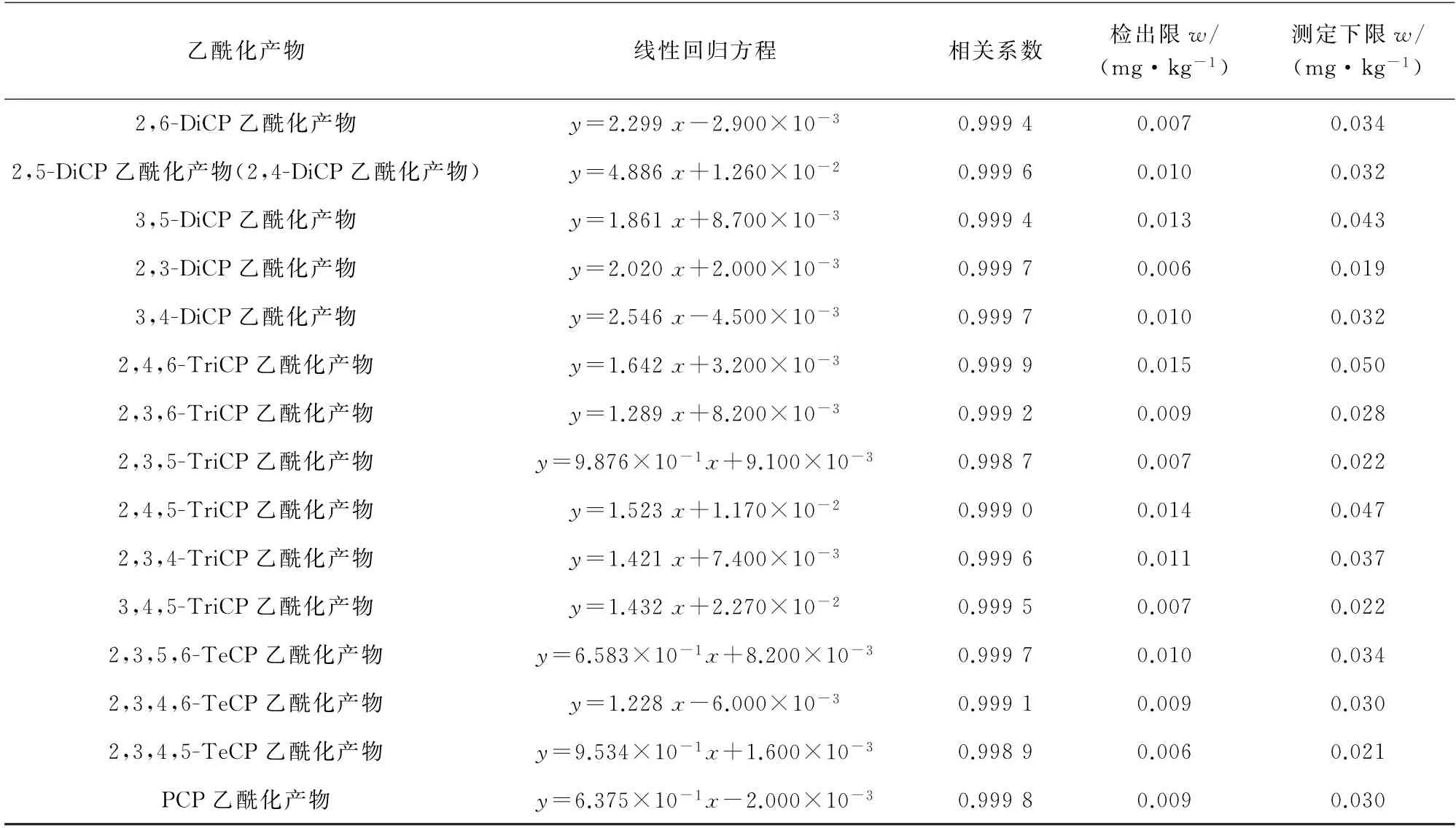
表2 (续)
2.6 回收和精密度试验
以空白皮革样品为基质,分别添加低、中、高等3个不同浓度水平的混合标准溶液,每个浓度水平制备6个平行样,按试验方法进行测试,计算回收率和测定值的相对标准偏差(RSD),精密度和回收试验结果见表3。
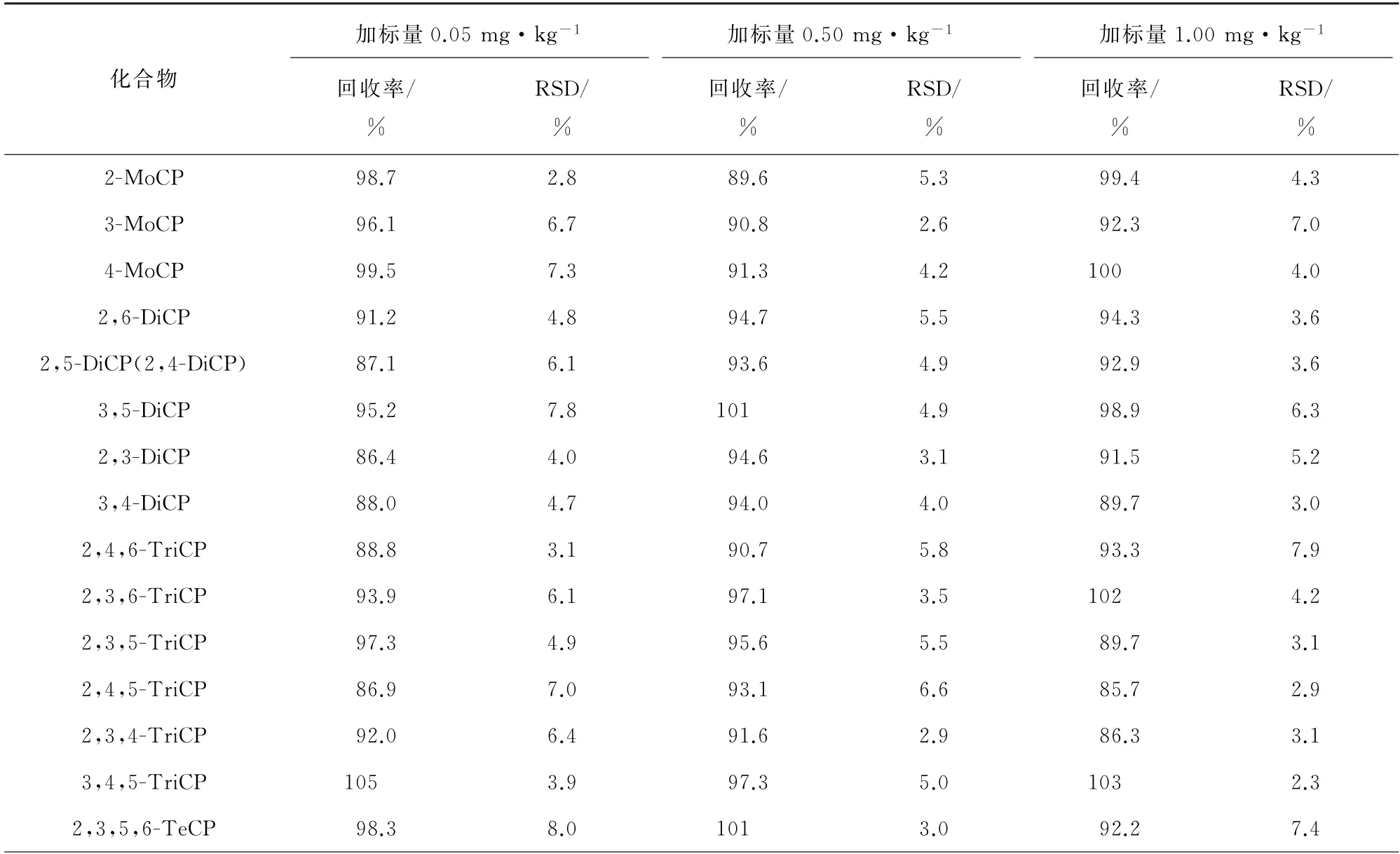
表3 精密度和回收试验结果(n=6)Tab.3 Results of tests for precision and recovery(n=6)
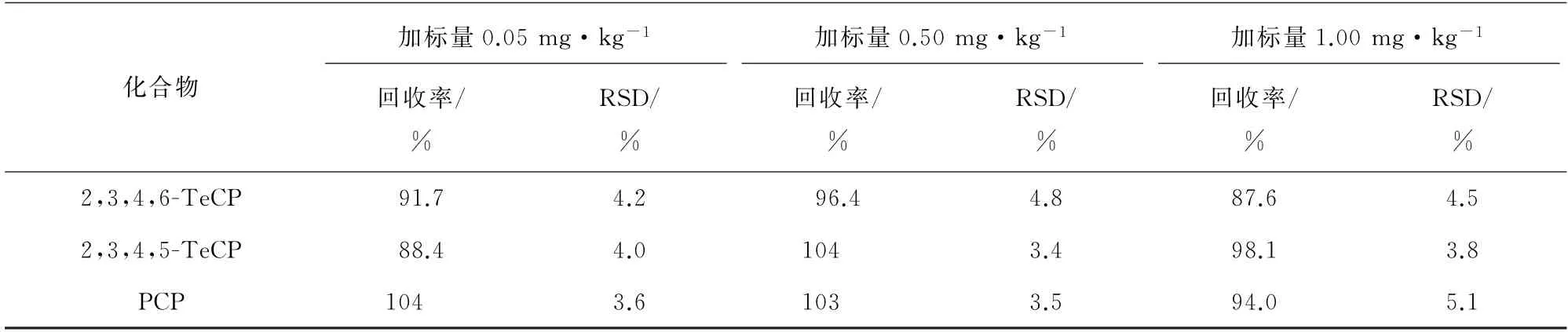
表3 (续)
由表3可知:19种含氯苯酚的回收率为85.7%~105%,说明该方法准确可靠;RSD为2.3%~8.0%,表明方法精密度较好。
2.7 样品分析
应用本方法分别对市售鞋材上取下的20个皮革样品进行分析,在5个样品中检出3,5-DiCP、2,4,6-TriCP、2,3,4,6-TeCP和PCP。其中,2,3,4,6-TeCP检出4次,2,4,6-TriCP检出3次,PCP检出2次,3,5-DiCP检出1次。最小检出值为0.20 mg·kg-1(2,4,6-TriCP),最大检出值为1.11 mg·kg-1(3,5-DiCP),共有5个检出值大于GB/T 18885-2009规定的限值(0.5 mg·kg-1)。
本工作建立了水蒸气蒸馏-气相色谱-质谱法测定皮革制品中19种含氯苯酚含量的方法。该方法具有提取效率高,样品提取液干净,目标峰的分离度好、准确度高、精密度好等特点,可为皮革制品中含氯苯酚化合物的测定提供了一种可靠实用的方法。
猜你喜欢
科学大众(2022年23期)2023-01-30 07:04:00
中国生物化学与分子生物学报(2022年8期)2022-09-08 00:39:50
中学生数理化·自主招生(2022年4期)2022-05-09 22:00:23
中国果业信息(2021年5期)2021-12-05 22:10:28
恋爱婚姻家庭·养生版(2020年11期)2020-12-17 03:26:48
红蜻蜓·低年级(2017年10期)2017-11-21 20:03:39
中国组织化学与细胞化学杂志(2016年4期)2016-02-27 11:15:53
合成化学(2015年10期)2016-01-17 08:56:24
中国当代医药(2015年16期)2015-03-01 02:03:13
小朋友·快乐手工(2014年7期)2014-08-18 12:22:07
