
紫外光本体法聚丙烯酸酯压敏胶的制备与表征*
2020-11-13 04:19周惠敏谢延林刘迎春朱梦璐王柱祥瞿雄伟
化学与粘合 2020年5期
周惠敏,谢延林,刘迎春,朱梦璐,王柱祥,瞿雄伟
(1.河北工业大学 化工学院,天津300130;2.河北冀工胶管有限公司,河北 衡水053200;3.京华塑业有限公司,河北 廊坊065800)
前 言
压敏胶粘剂PSAs(Pressure-sensitive adhesive)是指仅采用指压即可使胶粘剂立即与被粘物光洁表面粘接的一类胶粘剂[1]。丙烯酸酯压敏胶有多种分类方法,按照生产工艺的不同分为:溶液型、乳液型、热熔型和辐射型四种[2]。由于紫外光(UV)辐照技术具有引发效率高、可常温操作、节能和制备成本低、不含有机溶剂等特点,已被广泛应用于涂料、胶粘剂、油墨等多种领域[3~4]。本文采用丙烯酸酯作为原料,以紫外光引发聚合制备了具有一定黏度的预聚物,随后采用光固化方法,获得了聚丙烯酸酯压敏胶,考察了HDDA 交联剂用量对压敏胶性能的影响。
1 实验部分
1.1 原材料
丙烯酸正丁酯(BA):分析纯,普莱华化学试剂厂;丙烯酸羟乙酯(HEA)、丙烯酸(AA):分析纯,上海麦克林试剂有限公司;二苯基(2,4,6-三甲基苯甲酰基)氧化膦(TPO)、2-羟基-2-甲基-1- 苯基-1-丙酮(1173)和1,6-己二醇二丙烯酸酯(HDDA):分析纯,上海麦克林生化科技有限公司;四氢呋喃(THF)和甲苯:分析纯,天津福晨试剂厂。
1.2 实验步骤
1.2.1 丙烯酸酯预聚物的制备
将单体混合物BA(82.8g)、HEA(10g)和AA(7.2g)及0.2g TPO 光引发剂加入到装有冷凝和机械搅拌装置的四口烧瓶中。反应体系通氮气除氧后,用UVLED(TY-UV-8,广东东莞拓翼电子有限公司)(395nm)照射一定时间;当体系黏度增加到1200mPa·s-1,关闭光源得到丙烯酸酯预聚物。
1.2.2 压敏胶带薄膜的制备
在UV 辐照合成的丙烯酸酯预聚物中加入不同质量比的1,6-己二醇二丙烯酸酯(HDDA),并补加光引发剂TPO(1wt%)和1173(1wt%),搅拌均匀后脱泡。使用100μm 涂布器在50μm 厚的聚对苯二甲酸乙二醇酯(PET)薄膜上涂布,并控制胶层厚度为100μm。再用高压汞灯(XP-8,365nm,飞利浦仪器有限公司)照射20s,得到压敏胶带。
1.3 测试与表征
UV 引发制备预聚物的黏度按照GB2794-1995标准、使用旋转黏度计(NDJ-8S,上海方瑞仪器有限公司)测试;预聚物和交联聚合物的结构采用傅里叶变换红外光谱仪(Vector-22,德国Bruck 公司)表征,扫描范围为400~4000cm-1,分辨率为4cm-1;交联聚合物的凝胶含量由索氏提取装置在沸腾的THF中抽提24h 获得;其可溶部分相对分子质量用凝胶渗透色谱仪(PL-GPC220,英国Polymer Laboratories)测试,流动相为色谱级THF;其动态力学性能由英国Triton 公司的动态力学分析仪(Tritec2000,英国Triton 公司)在氮气氛测得,温度范围-70~80℃,升温速度为5℃/min,频率为1Hz;压敏胶样品由天津市材料实验机厂的涂膜涂布器(OTG 型)制备;压敏胶的性能包括垂环初黏力、180°剥离力和持黏性能按照FINAT Testing1,9,8 标准测试,测试温度23±2℃,相对湿度50±5%。
2 结果与讨论
2.1 丙烯酸酯聚合物的结构
2.1.1 红外光谱分析
图1 中为不同光照时间下丙烯酸酯聚合物的FTIR 图,其中,曲线a、b 和c 为加入0.9wt%HDDA交联剂的红外光谱,曲线d 为预聚物的红外光谱。由图1 可知,不同辐照时间下样品的红外谱图是相似的,C=C 双键吸收峰强度的变化表明了UV 固化过程的发生,1639cm-1和810cm-1处观察到的尖峰可归因于C=C 键的强吸收,随着辐照时间的增加,吸收峰强度降低,表明C=C 键参与反应。当固化时间达到20s 时,C=C 吸收峰消失,表明所有单体都反应完全。

图1 不同固化时间丙烯酸酯聚合物的红外光谱图Fig.1 The FTIR spectra of acrylate polymers with different curing times
2.1.2 交联聚合物凝胶含量和相对分子质量
常用凝胶含量和交联点间平均相对分子质量(Mc)来表征聚合物的交联程度,其取决于交联剂的含量和交联剂的官能团数。
聚合物交联点间的平均相对分子质量依据Flory-Rehner 方程计算[5]:

式中:V1为溶剂甲苯的物质的量体积(106.3cm3/mol)[6],ρp为产物的密度,可认为与聚丙烯酸丁酯的密度相同,为ρPBA(1.06g/cm3)[7],Φ 为溶胀于甲苯中交联聚合物的体积分数,可由下式计算得到:

式中:Wp和Ws分别为聚合物和甲苯的质量分数,ρp和ρs分别为聚合物和甲苯的密度(0.867g/cm3)。
χ为聚合物与甲苯的相互作用参数,可依据下式计算[8]:

式中:δ1和δ2分别为聚合物和溶剂甲苯的溶解度参数,因二者的溶解度参数相似[6],所以,χ=0.34。
由此可以得到聚丙烯酸酯交联点间的相对分子质量Mc,结果见表1。随着交联剂HDDA 含量由0wt%增加到0.9wt%,交联点间的相对分子质量由2.60104g/mol 降低到0.19104g/mol。
而由索氏提取器得到的可溶部分聚丙烯酸酯产物,可由GPC 测试得到其数均相对分子质量(Mn)、重均相对分子质量(Mw)和相对分子质量分布指数(MWD),结果也列于表1。由表1 可知,随着HDDA 交联剂含量的增加,产物的凝胶含量在增加。当不加交联剂时,凝胶含量为44.66%,这部分凝胶的产生是通过分子间或分子内的链转移,形成支链自由基,支链自由基间彼此偶合形成凝胶[9~10]。随着HDDA 含量的增加,大分子链上双键含量占比越大,形成的交联点越多,因此,压敏胶的凝胶含量越高。此时,交联点间的相对分子质量和可溶部分聚合物的相对分子质量如Mn、Mw都同时降低。

表1 聚合物相对分子质量参数汇总Table 1 The summary of the molecular weight parameters of the polymers with different HDDA contents
2.1.3 黏弹性
动态力学分析(DMA)反映了黏弹性材料在周期性交变应力作用下的分子运动情况。在外力作用下,聚合物材料的形变介于弹性材料和黏性材料之间,材料同时具有固体弹性和液体黏性的行为被称为黏弹性。聚合物的黏弹性对压敏胶的性能具有重要的影响。不同HDDA 交联剂含量的聚丙烯酸酯压敏胶的损耗因子(tanδ)和储能模量(G')随温度的变化如图2 所示。
由图2(a)可知,所有曲线仅存在一个玻璃化温度转变峰,并且峰宽变化不大,这表明,在不同HDDA 含量下,形成的聚合物为无规共聚物,且聚合物的玻璃化转变温度(Tg)随着HDDA 含量的增加而增加,见表1。由图2(b)可知,随着HDDA 含量的增多,聚合物在橡胶平台区的剪切储能模量在变大。这是由于随着HDDA 含量的增多,提高了聚合物的交联密度,更加致密的交联网络限制了线性分子的运动,聚合物的Tg 提高;同时,随着交联剂增加,聚合物分子链刚性变大,材料在外力作用下抵抗形变的能力提高,因此,模量更高。
聚合物的缠结相对分子质量(Me)也是影响压敏性能的重要因素,可由下式计算[11]:

其中ρP是指聚合物的密度,G0n是橡胶平台开始时的模量,T 为模量最低点的温度值,随着交联剂的加入,Me值明显降低,见表1。

图2 不同HDDA 含量的聚合物的动态力学曲线(a)tanδ-T,(b)G’-TFig.2 The DMA curves of polymers with different HDDA contents(a)Tanδ-T and(b)G,-T
因此,可用图3 来示意UV 引发、交联丙烯酸酯本体聚合反应过程。
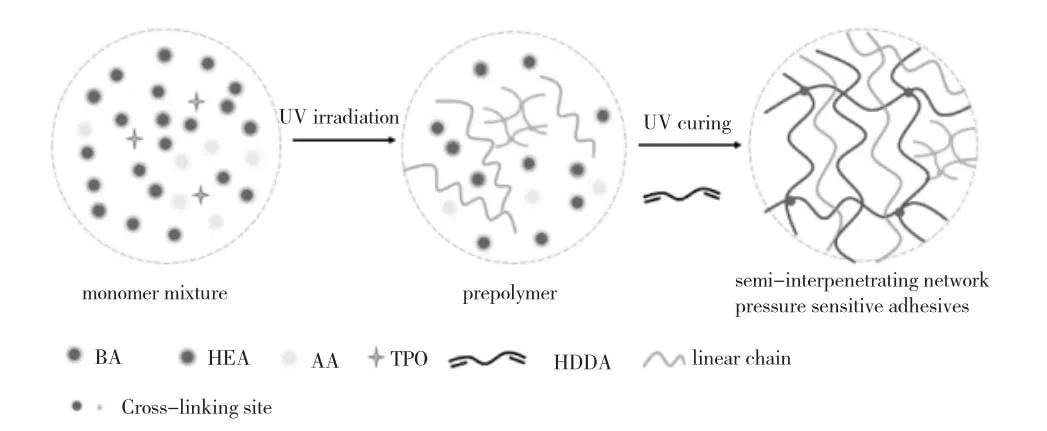
图3 本体法、UV 引发和交联丙烯酸酯聚合过程及其结构示意图Fig. 3 The schematic diagram of polyacrylate prepared via a bulk polymerization process triggered by a radical photo-initiator under UV irradiation and UV-crosslinking
2.2 压敏性能
聚丙烯酸酯压敏胶的压敏性能通常由初黏力、剥离力和持黏力三个值来评价。其压敏性能与聚合物的性质[12]、微结构[13]、黏附功[14]等有关。压敏性能与交联剂含量的关系列于表2 中。随着HDDA 含量的增加,初黏力和剥离力逐渐降低,而持黏性显著提高。
2.2.1 初黏力
压敏胶的初黏力与材料的界面作用力和本体压敏胶的流变性质有关[15],取决于变形和在较短接触时间粘合基底的能力。随着HDDA 含量的增多,初黏力在下降,这是由于聚合物内部凝胶含量的增加和Mc的减少,表明形成了较高交联密度的三维网络结构,导致聚合物链的形变能力降低,这进一步降低了PSAs 与基材之间的铺展、润湿、粘合。因此,随着交联剂HDDA 含量的增加,初黏力呈现下降的趋势。

表2 不同HDDA 含量对压敏胶性能的影响Table 2 The effect of HDDA contents on the properties of the pressure sensitive adhesive
2.2.2 180°剥离强度
剥离过程中测得的力由两部分组成:克服黏附功即破坏胶粘剂与基材界面结合所需的力;使胶粘剂本体发生变形,是黏弹性损耗能[16]。其也与相对分子质量,分子缠结网络和接触时间相关。由表2 可知,随着交联剂的增加,剥离力降低。这主要归因于两个方面。首先,交联剂含量的增加,使得聚合物交联密度增大,交联的网络结构限制了分子链的运动。其次。随着聚合物储能模量的增加,聚合物在剥离过程中丝状化现象减少,能量耗散较少[13]。
2.2.3 持黏性能
根据Dahlquist 方程,可知用剪切破坏时间表示的持黏性能与聚合物的零切黏度有关[18]:

式中,T 为剪切破坏发生的时间;L 是粘在被测表面的试样条的总长度;W 为试样条的宽度;η 为聚合物压敏胶的零切黏度;t 为胶层厚度;M 为测试载荷的质量;g 为重力加速度。该式应用于静态剪切试验,如果破坏发生的时间足够长,可认为最初的剪切速率很小并且滑移完全是由聚合物稳态黏性流动导致的。而观察到的所有试样的破坏方式都是内聚破坏,也可以说明这种假设与实际情况相符。从式(5)中可以看出,在其他条件不变的情况下,剪切破坏时间受聚合物零切黏度η 影响,即剪切破坏时间归因于凝胶含量的增加和交联网络更致密两个方面[19]。由表1 的结果,可以清楚地看到,随着交联剂HDDA 含量的增加,交联度增加,交联点间相对分子质量减小,缠结相对分子质量减小,可溶部分的重均相对分子质量是增加的,因此,持黏性能由1200min 增加到7400min,见表2。
3 结 论
通过UV LED 光聚合和与非共轭双烯烃丙烯酸酯单体(HDDA)用UV 固化交联方式制备了聚丙烯酸酯压敏胶。可以通过调控HDDA 交联剂含量来获得初黏力、剥离力和持黏性能三者间的平衡,制备性能优异的压敏胶。
猜你喜欢
辽宁化工(2022年5期)2022-05-28
精细石油化工(2022年3期)2022-05-27
粘接(2021年2期)2021-06-10
科学技术与工程(2020年3期)2020-04-08
当代陕西(2019年14期)2019-08-26
传媒评论(2019年4期)2019-07-13
民用飞机设计与研究(2019年4期)2019-05-21
华人时刊(2017年17期)2017-11-09
科学与财富(2017年12期)2017-05-16
科技创新与应用(2017年9期)2017-04-26
