
Ancient DNA molecular identification and phylogenetic analysis of Cervinae subfossils from Northeast China
2020-11-11 12:59:44XIAOBoSHENGGuiLianYUANJunXiaWANGSiRenHUJiaMingCHENShunGangJIHaiLongHOUXinDongLAIXuLong
古脊椎动物学报(中英文) 2020年4期
XIAO Bo SHENG Gui-Lian,2* YUAN Jun-Xia WANG Si-Ren HU Jia-Ming CHEN Shun-Gang JI Hai-Long HOU Xin-Dong LAI Xu-Long
(1 School of Environmental Studies, China University of Geosciences (Wuhan) Wuhan 430078 mr.shaw@outlook.com* Corresponding author: glsheng@cug.edu.cn)
(2 State Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences (Wuhan) Wuhan 430078)
(3 Faculty of Materials Science and Chemistry, China University of Geosciences (Wuhan) Wuhan 430078)
(4 Daqing Museum Daqing, Heilongjiang 163319)
(5 Paleontological Fossil Conservation Center, Qinggang County Qinggang, Heilongjiang 151600)
(6 School of Earth Sciences, China University of Geosciences (Wuhan) Wuhan 430074)
Abstract The deer resources in China are abundant, with seven species in the sub-family Cervinae distributing in various areas. The intraspecific phylogeny of Cervinae has been widely explored, while the evolutionary relationship among different species requires further efforts, in which only few molecular studies on ancient materials have been performed. In this study, we carried out ancient DNA research on two Cervinae subfossils from northeastern China, dating of 3800 and 5100 aBP. Through ancient DNA extraction, double-stranded sequencing libraries construction, next-generation sequencing and bioinformatics data analysis, we reconstructed two mitochondria sequences with lengths of 16475 bp (GenBank accession number: MT784751,sequence integrity: 99.83%) and 16167 bp (GenBank accession numberh: MT784752, sequence integrity: 97.96%), respectively. Based on the mitochondrial homologous sequences of the extant Cervinae species in GenBank, we constructed a phylogenetic tree. The results show that: 1) both the average length and the C-to-T substitution frequencies at 5’- end of the NGS short reads indicate the data are from ancient specimens; 2) the two ancient individuals clustered with Cervus elaphus in the phylogenetic tree, and were molecularly identified as C. elaphus; 3) the two ancient samples from Heilongjiang are phylogenetically close to the extant C. elaphus alxaicus, but far from the extant C. elaphus xanthopygus. Combining the dates of the samples, we suggest that these two samples represent a population of ancient C. elaphus in Heilongjiang, which was not the direct maternal ancestor of the extant C. elaphus xanthopygus.
Key words Heilongjiang, China; Cervinae; ancient DNA; molecular identification; phylogenetic analysis
鹿亚科(Cervinae)又称真鹿亚科,共分为4个属:鹿属(Cervus)、黇鹿属(Dama)、花鹿属(Axis)和麋鹿属(Elaphurus) (Sheng, 1992)。我国鹿类资源较为丰富,鹿亚科动物就包括鹿属的梅花鹿(Cervus nippon)、马鹿(C. elaphus)、坡鹿(C. eldi)、水鹿(C. unicolor)和白唇鹿(C. albirostris), 花鹿属的豚鹿(Axis porcinus)和麋鹿属的麋鹿(Elaphurus davidianus)共7个种(Tu et al., 2012)。中国现存的鹿种能表现出该亚科动物几乎全部系统发生的阶段,因此中国也被称为“鹿类动物进化的展览场”(Sheng, 1992)。
过去,人们对中国鹿类动物起源和系统进化及分类研究主要依据形态学特征,例如角的有无和形态、体型和体重、头骨和炮骨、泪窝和腺体的有无及特征、齿式和毛色特征、行为特征等,其中颅骨衍生物(牙齿、角等)是形态学系统分类的重要特征之一(Groves and Grubb, 1987; Sheng, 1992; Cai and Yin, 1992; Dong and Li, 2009; Dong et al.,2018)。基于化石的形态学研究是对古代鹿亚科动物研究的方法之一,盛和林(1992)认为鹿类动物的化石记录虽能较真实地反映其进化历史,但由于化石的分类基准大都使用角或容易变异的骨的形态,因此容易造成异议。Geist (1998)指出,偶蹄类的比较形态学在分类和进化研究中并不总是可靠的,因为这些动物的形态学特征随着环境条件变化而产生很大差异,所以结果不能被普遍接受。Emerson and Tate (1993)指出鹿类动物形态学研究所依据的角形和皮毛颜色等特征容易受到趋同或平行进化的影响。因此鹿科动物的形态学分类仍然存在较大争议。近40年来,很多学者在传统分类基础上引入了细胞学(Wang and Du, 1983; Neitzel, 1987)和分子生物学以及计算机技术对鹿类动物进行分类研究,从细节上对其进行了补充和修改。然而,涉及豚鹿、麋鹿和马鹿的系统分类地位,不同学者的观点尚存争议(Emerson and Tate, 1993; Grubb, 1993; Geist, 1998; Polziehn and Strobeck, 2002; Liu et al., 2003)。
古DNA是残存在古生物化石或亚化石、考古材料中的生物大分子,能从分子水平反映生物个体之间的遗传组成差异。发展利用古代鹿科动物的古DNA线粒体甚至核基因组信息,并结合现生鹿科动物的遗传信息和动物谱系地理学,综合分析研究中国鹿科动物系统发育是一个良好的发展方向(Liu et al., 2017)。本研究对采自黑龙江省肇东市和宾县的两个鹿科动物遗骨进行古DNA提取、DNA双链文库构建和高通量测序,通过生物信息学方法对序列数据进行处理,从分子水平对亚化石材料进行物种鉴定,结合现生部分鹿科动物基因序列,比较其同源序列之间的差异,通过系统发育分析探讨马鹿亚种间的进化关系。
1 材料与方法
1.1 实验材料
两个实验材料皆为黑龙江省大庆博物馆馆藏标本。据博物馆资料记载,CADG522于2008年发现于黑龙江省肇东市太平乡段松花江古河道;CADG527于2003年发现于黑龙江省宾县鸟河乡的泥沟。样品年代由美国BETA实验室通过放射碳同位素测定方法获得,相关信息如表1、图1所示。

表1 研究样品信息Table 1 Samples information
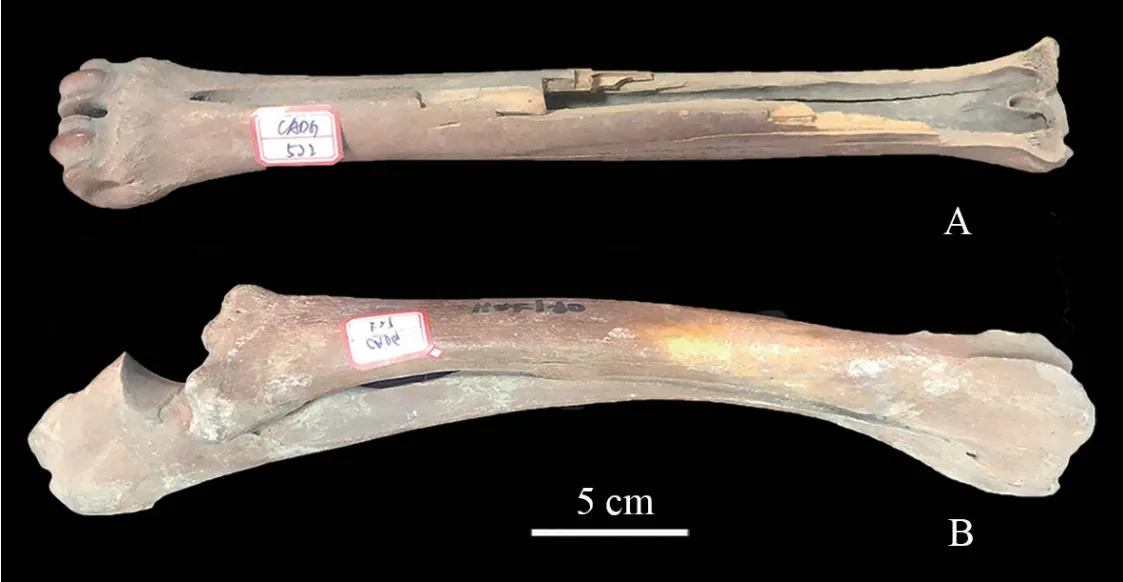
图1 产自黑龙江肇东太平乡(A)和宾县鸟河乡(B)的鹿类研究样品Fig. 1 Samples of Cervus sp. from Taiping, Zhaodong (A) and Niaohe, Binxian (B), Heilongjiang
1.2 古DNA提取
首先用4.5%的次氯酸溶液擦拭骨骼样品表面,再用无水乙醇擦拭两次并风干。使用切割机切取约300 mg骨骼样本,用已灭菌研钵研磨成粉末。按Rohland and Hofreiter(2007a, b)的方法,使用含EDTA (0.465 mol/L)和蛋白酶K (0.4 g/L)的裂解液充分裂解样品,再使用DNA纯化试剂盒(QIAquick PCR Purification Kit)结合超滤管(Millipore)完成样品古DNA的提取。
1.3 古DNA双链文库构建与测序
古DNA双链文库构建包括4个步骤(Matthias and Martin, 2010): 1) 末端修复(bluntend repair), 使用T4多聚核苷酸激酶(T4 polynucleotide kinase)和T4 DNA聚合酶(T4 polymerase)将DNA分子片段末端修复平齐;2) 接头连接(adapter ligation), 用快速连接酶(quick ligase)将接头(adapter)片段连接至修复平齐的双链DNA分子末端;3) 接头补齐(adapter fill-in), 使用Bst DNA聚合酶将连接接头的黏性末端补齐为平末端;4) 文库分子标记(indexing), 通过添加不同的小片段序列对DNA分子进行特异性标记,以在混合文库测序时对单个文库进行区分,该过程主要使用的酶为Q5高保真DNA聚合酶(high-fidelity DNA polymerase)。最后对构建的文库进行一次磁珠纯化即可得到用于测序的古DNA双链文库。构建完成的文库送北京泛生子基因测序公司进行双向测序。本研究对两个样品分两个批次各构建两个文库(522-1, 522-2及527-1, 527-2), 对第一批次两个文库进行预测序得到测序数据522-1Y和527-1Y, 深度测序得到测序数据522-1S和527-1S。第二批次文库直接进行深度测序得到测序数据522-2S和527-2S。
1.4 测序数据处理与分析
测序公司提供的测序序列文件为FASTQ格式,对每一样品的每一文库,分别使用“cutadapt-1.12” (Martin, 2011)切除接头序列并过滤掉低于30 bp的短片段,然后用软件“BWA-0.7.15” (Burrows-Wheeler Alignment) (Li and Durbin, 2010)中的“aln”和“samse”将片段与参考基因组比对,舍弃比对质量得分低于30的序列;运用SAMtools v1.3.1软件(Li et al., 2009)中的“view”和“sort”算法将剩余序列按5’端位置在参考基因组上进行排序,用“rmdup”去除可能存在的PCR重复序列,用“merge”算法得到每一文库的单一bam格式文件。将同一样品的不同文库bam文件合并,再运行“rmdup”去除可能存在的重复序列,得到同一样品的合并bam文件,最终用angsd-0.916软件(Korneliussen et al., 2014)“-doFasta”导出匹配得到的各样品基因序列FASTA格式文件。
对古DNA分子碱基损伤的评估,使用各样品不同文库处理合并、并去掉重复后的bam文件作为输入文件,使用“mapDamage”软件包(Aurelien et al., 2001)统计片段末端5’端和3’端碱基损伤。
序列匹配时,先以麋鹿线粒体基因组序列(GenBank收录号:NC018358)作为参考序列对测序数据进行初步比对,将所得线粒体序列数据导出,使用BLAST (https://blast.ncbi.nlm.nih.gov)在线检索,得到样品序列的物种信息,初步判定实验样品的种属类型。本研究中对两个样品的第一批次得到的第一个文库,先后使用麋鹿(GenBank登录号:NC018358)和马鹿阿拉善亚种(GenBank登录号:KU942399)的线粒体基因组作为参考基因组对预测序数据(522-1Y和527-1Y)进行匹配分析。得到第二批次的各两个文库的深度测序结果(522-1S, 522-2S和527-1S, 527-2S)后,只选用在第一个文库匹配分析中匹配度较高的参考基因组用作合并后的参考基因组,对两个样品各文库分别进行数据过滤和合并处理,最终得到本研究的线粒体古DNA序列。
1.5 系统发育分析
利用Bioedit 7.0.9.0 (Hall, 1999)对测得的序列进行排列对比并辅以人工校对,以所得两个样品的古DNA序列与36个鹿亚科动物和一个外类群驼鹿(Alces alces)的同源序列(15109 bp, 不包含D-loop区)在CIPRES网站(http://www.phylo.org)使用PartitionFinder2(Cognato and Vogler, 2001)和RAxML V8.0 (Alexandros, 2014)通过500次自举检验估计节点构建系统发育树,碱基替换模型(model for substitution)选择GTRCAT。系统发育树的展示输出通过Figtree v1.4.2 (Rambaut, 2009)完成。分析中所用序列信息见表2, 序列选取兼顾鹿亚科在中国分布的4属9种。同时,与样品聚类较近的中国马鹿5亚种和梅花鹿选取了当前GenBank中所收录的所有高质量序列。

表2 系统发育分析所使用序列信息Table 2 Sequence information used in phylogenetic analysis
2 结果
2.1 古代线粒体DNA片段末端碱基特性
运用mapDamage软件对两个样品所得线粒体DNA二代测序序列进行分析,末端碱基替换统计结果如图2所示。CADG522和CADG527在5’端的“C→T”碱基替换频率分别为6.883%和6.897%, 3’端的“G→A”碱基替换频率分别为6.105%和9.123%。
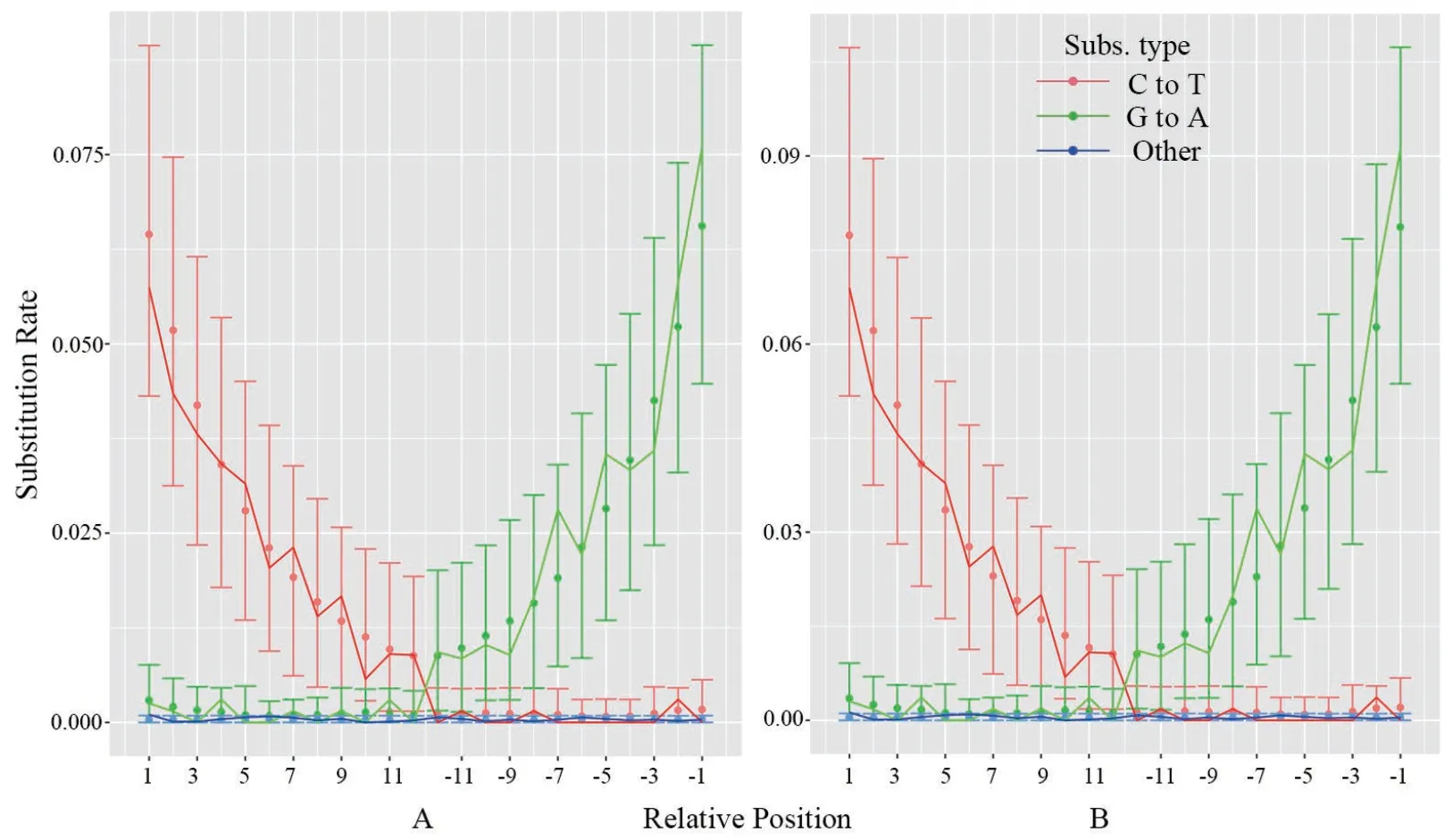
图2 样品CADG522 (A)和CADG527 (B)线粒体DNA片段末端核苷酸错配统计Fig. 2 Statistical analysis of nucleotide misincorporation observed on reads of samples CADG522 and CADG527
2.2 二代测序数据及序列比对结果
先后使用麋鹿(GenBank收录号:NC018358, 全长16355 bp)和马鹿阿拉善亚种(GenBank收录号:KU942399, 全长16503 bp)的线粒体基因组作为参考基因组,对两个样品第一批次的文库(522-1和527-1)的预测序数据(522-1Y和527-1Y)进行匹配分析,结果显示两个样品均与阿拉善马鹿匹配度较高。最终以阿拉善马鹿线粒体基因组作为参考基因组对文库深度测序数据(522-1S, 522-2S和527-1S, 527-2S)进行过滤、匹配和合并处理,分别得到两个样品16475 bp (GenBank收录号:MT784751, 序列完整度:99.83%)和16167 bp (GenBank收录号:MT784752, 序列完整度:97.96%)古DNA线粒体基因序列。文库测序信息如表3所示。
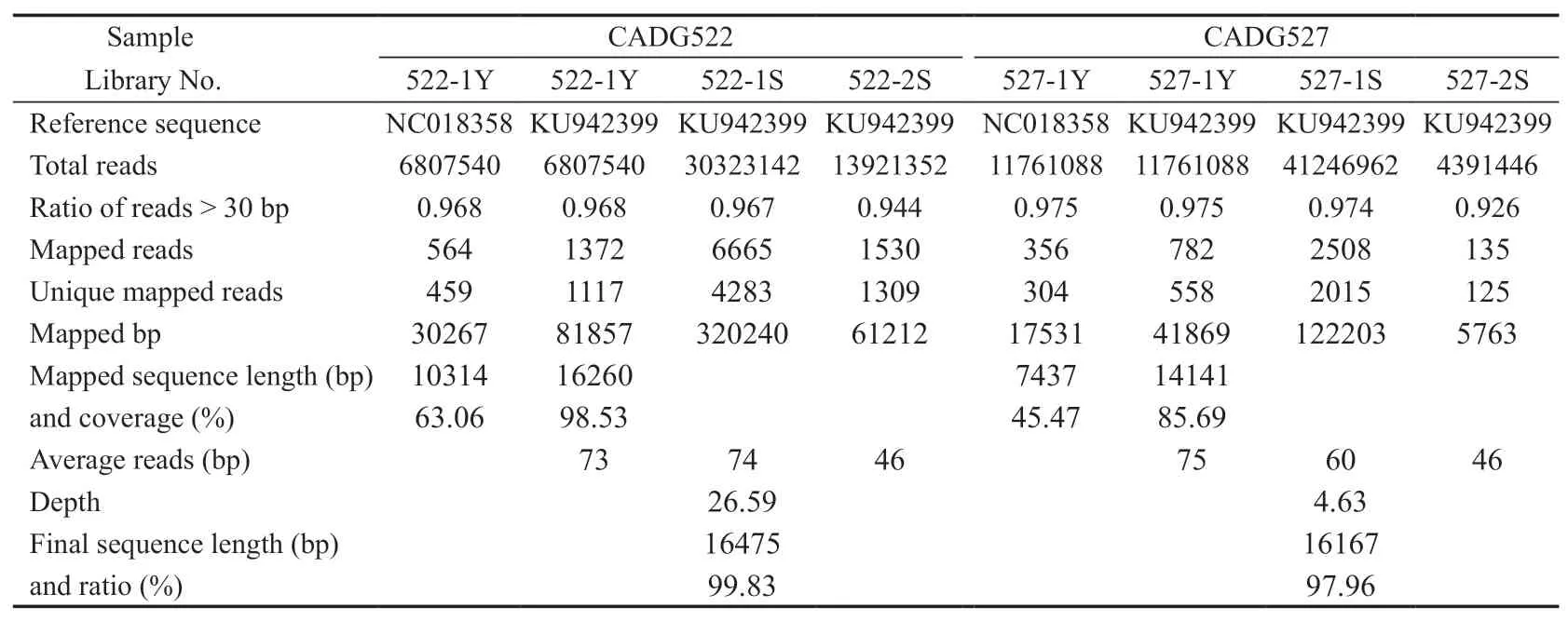
表3 二代测序数据信息Table 3 Next-generation sequencing data
2.3 系统发育树
基于本研究所得两条古DNA线粒体序列和从GenBank检索所得36条鹿亚科动物的线粒体基因组序列,以驼鹿(A. alces)为外类群(outgroup)构建的分子系统发育树(图3)显示,两个古代样品(CADG522和CADG527)都与马鹿阿拉善亚种聚为一支,马鹿阿拉善亚种、天山亚种、甘肃亚种和东北亚种一起与梅花鹿互为姐妹群,且与后者一起共同构成马鹿塔里木亚种的姐妹群。白唇鹿两个个体聚为一支处于水鹿大分枝内部。坡鹿与3个麋鹿聚为同一分枝,往树根依次为黇鹿属的黇鹿和花鹿属的花鹿和豚鹿。
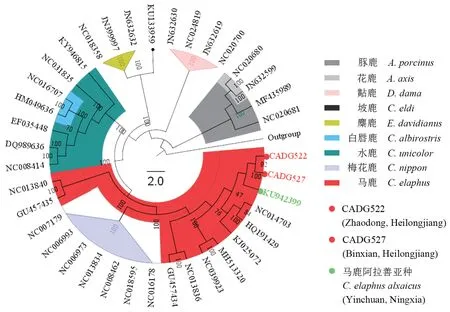
图3 鹿亚科线粒体基因组15109 bp同源序列ML法系统发育树Fig. 3 ML phylogenetic tree of Cervinae species based on 15109 bp mitochondrial genome homologous sequences
3 讨论
3.1 古代样品线粒体DNA真实性鉴定
本研究所得古DNA序列真实性可从如下方面论证:首先,采集样品时严格按照古DNA研究规范,样品的处理、提取及建库都在古DNA专用实验室的独立环境进行,实验者严格遵守古DNA实验防污染规则操作。其次,实验过程中,提取、建库全程设置有空白对照及正对照,对照实验组显示无异常,排除试剂、操作者及样品之间的交叉污染。再次,本研究所得序列经BLAST查询,所得推荐高可信度序列均为鹿科动物线粒体基因序列;本实验室从未进行现代鹿科动物分子生物学实验,所得序列确定来源于古代样品。最后,本研究各文库所得线粒体DNA片段平均长度较短,约45~75 bp, 符合古DNA片段特征(Hofreiter et al., 2014)。同时,经二代测序所得两个样品的DNA片段的5’端碱基“C→T”替换频率分别为6.883%和6.897%, 符合Sawyer et al. (2012)关于古代线粒体DNA片段5’端碱基“C→T”替换频率与样品年代的相关性研究结果。综上,本研究所得序列是来自古代样品的古DNA序列。
3.2 鹿亚科动物的系统演化关系
近年来,关于鹿亚科各属种的系统演化关系,前人的蛋白质电泳、染色体组型、线粒体控制区序列等研究表明麋鹿和豚鹿与鹿属动物的进化关系较近,认为麋鹿和豚鹿应该并入鹿属,归并后的鹿属为单系发生(Wang and Du, 1983; Randi et al., 2001; Liu et al., 2003)。本研究加入古代鹿科动物样品序列后构建的系统发育树(图3)显示,麋鹿与鹿属的坡鹿互为姐妹支,且自展支持率为100%, 支持Randi et al. (2001)将麋鹿归为鹿属的观点;但是豚鹿在系统发育树中的位置与鹿属各种距离较远,且豚鹿与花鹿归为同一分支,因此,本研究不支持王宗仁、杜若甫(1983)和刘向华等(2003)将豚鹿划归鹿属的观点。
在鹿属内部,王宗仁、杜若甫(1983)认为梅花鹿与马鹿的关系最近,现生鹿属动物中水鹿是最原始的,白唇鹿与梅花鹿几乎同时从水鹿的祖先种分化而来,马鹿则是最后分化出来的。图3中,梅花鹿并非直接从水鹿分化而来,梅花鹿分支与马鹿阿拉善亚种、天山亚种、甘肃亚种和东北亚种共同聚类形成的分支构成姊妹群,而马鹿塔里木亚种处于二者的根部。该结果显示马鹿塔里木亚种在早期从马鹿和梅花鹿的共同祖先分化而出,而后依次分化出梅花鹿和马鹿其他亚种,从线粒体基因组水平印证了王宗仁等的结论。
3.3 马鹿种群的基因交流
关于马鹿的系统进化关系,大多数观点认为马鹿分为东西两个进化种群:西部群体包括欧洲种群和我国新疆南部塔里木亚种,东部种群包括北美种群、亚洲及我国新疆北部的种群(Polziehn and Strobeck, 2002; Muhmut et al., 2002; Ludt et al., 2004)。本研究的系统进化树显示塔里木马鹿与其他生活在中国的4个马鹿亚种来源于共同祖先的两个不同分支,支持马鹿在进化中分出东西两个种群的观点。塔里木马鹿较早地分化出来,在一个封闭的地理环境下独立进化,因地理隔离而有别于其他马鹿种群。之后,东北亚种、甘肃亚种、天山亚种和阿拉善亚种相继分化,这也证明了中国马鹿是从中东和欧洲返回中国的过程中从西向东逐渐分化的(Sheng, 1992)。
现分布于宁夏和内蒙古交界的贺兰山中段的阿拉善马鹿种群,是我国唯一幸存的马鹿阿拉善亚种的有效种群(Li et al., 1998; Wang et al., 1999)。受贺兰山周围被城市、沙漠、河流隔断形成孤岛的地理特征所影响,该种群已成为一个隔离种(Qiao et al.,2019)。本研究涉及的两个古代鹿科动物样品,校正年代分别为(3800±30)和(5100±30)aBP, 采集地黑龙江肇东市和宾县与贺兰山相距约1900 km。黑龙江地区的古代样品在系统发育树上与贺兰山的现生马鹿阿拉善亚种聚为一支,而与现生马鹿东北亚种亲缘关系较远,表明在历史时期两地的马鹿种群存在较密切亲缘关系,可能发生过种群消退、迁徙、引入等,也可能曾有过高于现生种群的基因交流;两例古代样品代表的古代黑龙江马鹿种群,不是现生马鹿东北亚种的母系直系祖先。本研究后续工作将对不同历史时期、不同地点的马鹿古代样品的线粒体基因组及核基因组进行研究,以期为马鹿各亚种的种群演化历史提供更为全面的分子证据。
猜你喜欢
Natural Products and Bioprospecting(2023年6期)2023-12-29 13:42:30
热带亚热带植物学报(2022年4期)2022-08-18 00:36:20
——和田盘羊
野生动物学报(2021年4期)2021-11-14 06:13:24
猪业科学(2021年3期)2021-05-21 02:05:36
幽默大师(2020年10期)2020-11-10 09:07:22
中华诗词(2019年1期)2019-11-14 23:33:56
小哥白尼(野生动物)(2018年1期)2018-05-26 09:28:03
猪业科学(2018年4期)2018-05-19 02:04:31
小哥白尼·野生动物画报(2018年1期)2018-02-05 16:49:12
新疆大学学报(自然科学版)(中英文)(2016年4期)2016-05-16 05:56:12

- 古脊椎动物学报(中英文)的其它文章
- New skulls of ctenodactyloids from the Early Oligocene of Ulantatal, Nei Mongol, China
- Taxonomic revision of Anthracokeryx thailandicus Ducrocq, 1999 (Anthracotheriidae, Microbunodontinae)from the Upper Eocene of Thailand
- New chroniosuchian materials from Xinjiang, China
- A new species of Luganoia (Luganoiidae, Neopterygii)from the Middle Triassic Xingyi Biota, Guizhou, China
- Subdivision and age of the Silurian fish-bearing Kuanti Formation in Qujing, Yunnan Province
- 总 目 次