
Sympathetic nervous system activation and heart failure:Current state of evidence and the pathophysiology in the light of novel biomarkers
2020-11-05 00:21:22JosipAneloBorovacDomenicoAmarioJoskoBozicDuskaGlavas
World Journal of Cardiology 2020年8期
Josip Anđelo Borovac,Domenico D'Amario,Josko Bozic,Duska Glavas
Abstract
Key words:Autonomic nervous system;Biomarkers;Catecholamines;Catestatin;Chromaffin system;Epinephrine;Heart failure;Myocardial failure;Norepinephrine;Sympathetic nervous system
INTRODUCTION
Heart failure(HF)is a complex clinical syndrome characterized by the symptoms such as breathlessness,fatigue and ankle edema and signs like elevated jugular venous pressure,lung crepitations during auscultation and peripheral edema[1].The central hemodynamic consequence of HF is the inability of a heart to support required metabolic demands and perfusion of organs and tissues due to structural and/or functional cardiac abnormalities that predilect to decreased cardiac output(CO)and/or increased intracardiac filling pressures during the rest or exercise[2].HF nowadays represents a relevant clinical entity and global pandemic that affects more than 26 million adults worldwide while its general prevalence in population accrues to about 2% with yearly incidence of approximately 0.2% in Western countries[3].Projected burden of HF,assuming the stable incidence of this syndrome in persons ≥65 years was already surpassed by the actual burden of the HF in the United States and it is expected that more than 8 million people will have this condition in the United States by 2030[4,5].This increase in HF prevalence observed worldwide might be attributed not necessarily to increased HF incidence but to phenomena such as advancing age of the population and increased comorbidity burden coupled with improved HF survival due to progress in HF treatment and diagnosis while the decreased incidence of HF according to some data might partially be the consequence of more efficacious treatment of acute coronary syndromes,lower severity of index HF events and improvements in HF primary prevention programs[5,6].
Despite the advancements in therapeutic management,HF is still characterized by the rather high morbidity and mortality rates and considerable healthcare expenditures while these outcomes appear to be strongly dependent on the region of the world,healthcare infrastructure and the level of quality/access to specialized HF care[5,7-10].Of note,HF is at least as deadly or even deadlier than some of the common malignancies in both men and women.Among men,patients with HF had worse mortality outcomes than those with prostate and bladder cancer while among women those with HF had worse mortality outcomes than female patients suffering from breast cancer[11].Survival after a diagnosis of HF has shown only modest improvement in the 21stcentury,with an increase in average survival rates between 6.4% to 7.2%during nearly two decades thus clearly indicating that our clinical efforts to improve outcomes in HF substantially lag behind advancements in other severe conditions such as cancer[12].In support to this notion,a recent data from the United States nationwide temporal analysis showed that age-adjusted death rates for HF did not change significantly,in fact there even emerges a trend of the slight rise of HF-related mortality recently,after nearly 15 years of modest but gradual decline in HF-related mortality since the late nineties[13].Similarly,recent longitudinal data acquired from the Framingham Heart Study and Cardiovascular Health Study showed that HF incidence was relatively stable for almost two decades and this was true for mortality outcomes as well(including cardiac death,non-cardiac death,and all-cause mortality)[14].This study also showed that the incidence of heart failure with reduced ejection fraction(HFrEF)significantly declined whereas the incidence of heart failure with preserved ejection fraction(HFpEF)significantly increased over time in both sexes.Approximately 50% of community patients with HF nowadays have an HFpEF clinical phenotype while multimorbidity seems to be a stronger driver for HFpEF onset although it is a highly prevalent phenomenon in both HFrEF and HFpEF.Likewise,both phenotypes portend a comparable 5-year mortality[15-17].Finally,the proportion of those dying of non-cardiovascular causes seems to be higher in HFpEF than HFrEF and this holds for non-cardiovascular-related 30-d readmissions that are more common among HFpEF compared with HFrEF patients[15,18].
Taken together,these recent trends strongly suggest that HF is a clinical entity that will continue to impose a significant burden on modern societies,urging for the advances in our understanding of its complex pathophysiology and development of new treatments.Equally important,the discovery and implementation of biomarkers that might aid in the diagnosis,prognosis,and risk stratification of patients with HF is required in a contemporary clinical practice[19].For these reasons,aims of the present review are to provide recent updates regarding the HF pathophysiology with the special emphasis on novel biomarkers that might reflect the sympathetic nervous system(SNS)activation as one of the constituent neurohumoral pathways that are upregulated to preserve CO in the setting of a failing heart.
PATHOPHYSIOLOGY AND COMPENSATORY MECHANISMS IN HEART FAILURE
Any abnormality or combination of abnormalities that cause structural,mechanical,or electrical dysfunction of the heart carry the potential to induce HF.Most commonly HF is the consequence of the myocyte injury caused by coronary artery disease,uncontrolled arterial hypertension and diabetes mellitus,however,adverse myocardial remodeling can be triggered and sustained by valvular dysfunction,tachyarrhythmias(especially atrial fibrillation/flutter),interatrial and interventricular conduction abnormalities or pulmonary disorders such as chronic obstructive pulmonary disease or pulmonary arterial hypertension[20].Less common etiologies include cardiomyopathies,myocarditis,infections,systemic toxins,and cardiotoxic drugs that are nowadays increasingly used in various chemotherapeutic regimens[21,22].At least several pathophysiological mechanisms are at play in the setting of failing myocardium such as increased hemodynamic overload,ventricular dysfunction due to subclinical or overt ischemia,pathologic ventricular remodeling,upregulated neurohumoral activation,impaired intracellular calcium cycling and accelerated apoptosis of cardiac myocytes,imbalance in the formation and breakdown of the extracellular matrix,and various genetic predilections[2].
Clinically,a majority of patients with HF have both systolic and the diastolic dysfunction occurring at the same time and these two pathophysiological mechanisms often overlap but even in the isolation of each other,they cause a similar degree of HF signs and symptoms[23,24].For the didactic purposes,in the systolic dysfunction,the primary pathomorphological substrate is the loss of functional myocardium(primary myocyte injury)most commonly due to ischemic disease and myocardial infarction causing impaired contractility and insufficient emptying of the ventricles consequently leading to increased left ventricular(LV)end-diastolic and end-systolic volumes and rise in end-diastolic pressure(LVEDP)within the left ventricle further decreasing stroke volume and left ventricular ejection fraction(LVEF)[25,26].An increase in LVEDP might retrogradely increase left atrial(LA)pressure which consequently increases pressure in the pulmonary circulation,and if this cascade progresses even further can induce right heart failure,congestive hepatopathy and affect portal and peripheral circulation thus altogether precipitating fluid extravasation leading to pulmonary and/or splanchnic and peripheral congestion.
On the other hand,in diastolic dysfunction,the contractile ability of the heart might be preserved,however,functional mechanisms that are responsible for the adequate filling of the heart are impaired.It is estimated that up to 50% of patients presenting with signs and symptoms of HF have normal or near-normal LVEF but exhibit abnormalities predominantly in diastolic function[27,28].Even more,those with normal LVEF by conventional transthoracic echocardiography and verified diastolic dysfunction can often have subclinical contractile dysfunction that is captured only by the means of myocardial deformation studies such as LV global longitudinal strain and speckle tracking techniques or advanced cardiac imaging methods such as cardiac magnetic resonance[29-31].These filling abnormalities may occur due to impairments in early diastolic relaxation of the LV(an active energy-consuming process)and/or increased stiffness and rigidity of the LA and LV(a passive process independent of energy)coupled with reduced arterial compliance in both major vessels such as the aorta and peripheral arteries[32,33].Among patients with HFpEF both processes of active relaxation and increased passive stiffness are impaired and are predominant pathophysiological mechanisms leading to diastolic dysfunction[34,35].These abnormalities altogether act synergistically to produce a rise in the LVEDP thus causing significant venous congestion in HFpEF patients that is as severe as among those with HFrEF[36].Of note,significant increase in passive LV stiffness is propagated by aberancies in collagen-dependent and titin-dependent deposition cellular pathways[35].Similarly,longstanding elevated ventricular pressures further perpetuate LA dilation that is clinically detected as an increased LA volume at rest and reduced LA filling during submaximal exercise[37].Also,peripheral oxygen extraction is blunted in HFpEF resulting in exercise intolerance while reduced peak oxygen uptake and increased perfusion/ventilation mismatch carry important prognostic information and assist in the selection of patients that might require advanced HF interventions such as heart transplantation or deployment of ventricular assist devices[38-41].
A complex interaction of highly prevalent comorbidities such as salt-sensitive hypertension,obesity,diabetes mellitus,metabolic syndrome,iron deficiency,chronic obstructive pulmonary disease and,atrial fibrillation(AF),combined with natural pathophysiological effects of aging can give rise to systemic proinflammatory state that affects coronary microvasculature and endothelium by upregulating cytokinemediated inflammation pathways[42,43].In this proposed pathophysiologic scheme,pioneered by Paulus and Tschöpe[44]in 2013,endothelial inflammation of coronary microvasculature acts as a central transitioning mechanism by which synergistic effects of comorbidities are translated onto heart thus causing secondary myocyte injury that ultimately leads to structural and functional alterations of the myocardium in HFpEF[44].According to the postulated model,coronary microvascular endothelial inflammation reduces nitric oxide(NO)bioavailability and cyclic guanosine monophosphate(cGMP)content and reduces protein kinase G activity in adjacent cardiomyocytes thus highlighting NO-cGMP-PKG signaling pathway disruption as the key culprit in HFpEF pathophysiology.This disruption leads to the onset of cardiac hypertrophy and increased resting tension(Fpassive)of cardiomyocytes due to hypophosphorylation of titin and increased myocardial nitrosative/oxidative stress[45-47].Furthermore,hypophosphorylation of constitutive myofilament proteins and increased calcium sensitivity of sarcomeres causes increased LV stiffness and abnormal relaxation contributing to HFpEF onset while these derangements are not present in normal myocardium[48].Grazianiet al[49]also proposed that microvascular dysfunction is the common pathophysiological pathway contributing to both microvascular angina and HFpEF[49].Of note,endothelial dysfunction represents a pathological vascular phenotype of all systemic arteries that encompasses damaging effects of vasoconstrictive,prothrombotic and proinflammatory substances and mediators on the endothelial vascular lining and diminished repairability of endothelium thus further acting as an independent pathobiological driver of atherosclerosis and overt cardiovascular disease[50-52].
Furthermore,a novel pathophysiological concept of endothelial-to-mesenchymal transition has been recently proposed describing a process by which endothelial cells undergo a series of molecular events that lead to a loss of their endothelial properties and a consequent shift in phenotype toward mesenchymal cells such as myofibroblasts,smooth muscle cells,and osteoblasts[53].Accumulation of these cells promotes plaque formation and atherosclerosis by secreting proinflammatory cytokines and metalloproteinases and increasing extracellular matrix and collagen deposition thereby affecting the structure and function of cardiac valves,native vein grafts that are used in coronary artery bypass graft surgery and inducing interstitial cardiac fibrosis,diastolic dysfunction,endocardial fibroelastosis and contributing even to the development of pulmonary arterial hypertension[54-59].From the molecular perspective,it seems that activation of transforming growth factor-beta plays a key role in the initiation of endothelial-to-mesenchymal transition cascade and tissue fibrosis through its interaction with SMAD-2/3/4 and SLUG signaling pathways[57,60,61].Furthermore,endothelial cells in which the EndoMT pathway was experimentally activated had significantly elevated secretion of proinflammatory cytokines such as interleukin-6,interleukin-8 and tumor necrosis factor-alpha thus likely representing an integrative pathophysiological cross-talk between fibrosis and inflammation[59,62].In summary,EndoMT might be the key link in interaction between inflammation,endothelial dysfunction,and chronic cardiac fibrosis,and thus might become a viable target for novel therapeutic solutions for cardiovascular disease[63,64].Altogether,these converging and mutually complementary pathophysiological mechanisms may contribute to a net effect of stiffening of cardiac myocytes and overt interstitial fibrosis thus directly inducing myocardial dysfunction during diastole and subsequent HF development.
In order to maintain adequate tissue perfusion in the setting of the failing heart,several compensatory mechanisms are activated to increase COviathe Frank-Starling mechanism,increased ventricular volume and wall thickness through the process of ventricular remodeling and augmenting mean arterial pressure(MAP)by activating several neurohormonal pathways and cytokine systems[21].These compensatory mechanisms are initially able to compensate for impaired myocardial function,however,they inflict deleterious effects on cardiac structure and function if chronically activated leading to further worsening of HF and progressive clinical deterioration of a patient.Neurohumoral systems that are upregulated act to promote beneficial shortterm changes in heart,kidneys,and vasculature to maintain cardiovascular homeostasis[65].They encompass the activation of the renin-angiotensin-aldosterone system(RAAS),arginine-vasopressin(antidiuretic)system,kallikrein-kininogen-kinin system,activation of natriuretic peptides system,neprilysin signaling pathway,endothelin pathway,and cytokine systems[66-70].Finally,the upregulation of adrenergic/SNS pathways and blunted responsiveness of the parasympathetic nervous system(PNS),also collectively known as autonomic nervous system(ANS)imbalance,is one of the hallmark neurohumoral disturbances that are operative in HF and is of central interest in this review[71,72].The summary of the most common etiologies,pathophysiological effects,and compensatory mechanisms in HF is shown in Figure 1.
It is worth of brief mentioning that evidence-based treatments such as angiotensinconverting enzyme(ACE)inhibitors,angiotensin receptor blockers,beta-blockers,mineralocorticoid receptor antagonists,angiotensin receptor neprilysin inhibitors,ivabradine,sodium/glucose cotransporter 2 inhibitors,digoxin,and cardiac resynchronization therapy(CRT)devices are developed around our understanding of compensatory and maladaptive mechanisms in HF[1,73].Importantly,these treatment modalities were found successful in reducing mortality rates and hospitalizations in patients with HFrEF,however,no evidence-based pharmacologic treatments with clear beneficial effects on these endpoints were observed in patients with HFpEF while current guidelines stipulate symptom control with diuretics and efficacious management of comorbidities such as arterial hypertension,AF,obesity,and diabetes in this population[1,74].
Recent European Society of Cardiology and Heart Failure Association expert panel issued a scientific position statement in which ANS imbalance is recognized as an important contributor to cardiac disease progression and is designated as a prognostic parameter and a therapeutic target in HF by the means of novel pharmacologic and/or device therapies[75].Furthermore,heart and brain are in bidirectional interaction meaning that depressed cardiac function affects cerebral structures and functional capacity while dysregulation of neuro-cardiac reflexes significantly affects the cardiovascular system thus aggravating and further sustaining the progression of HF[76].
PHYSIOLOGY OF SYMPATHETIC NERVOUS SYSTEM AND ITS MEDIATORS
In the advent of our understanding of HF,this syndrome was largely perceived as a hemodynamic disorder thus all treatment strategies were primarily directed toward the correction of hemodynamic abnormalities.However,since hemodynamic derangements could not fully explain the progression and long-term effects of the disease,a neurohormonal hypothesis was developed in which neurohumoral mechanisms encompassing RAAS and SNS activation were emphasized as independent drivers of cardiac dysfunction and progression of HF[77].
SNS activation is a fundamental physiological response to stress conditions(also known as the fight-or-flight response)such as hypovolemia,hypoglycemia,hypoxia or cardiovascular dysfunction[78].SNS activity can modify and induce a wide spectrum of potent hemodynamic effects such as an increase in heart rate(positive chronotropic effect),augmentation of cardiac contractility(positive inotropic effect),accelerated cardiac relaxation(positive lusitropy),enhanced(shortened)atrioventricular conduction(positive dromotropy),reduced venous capacitance and peripheral vasoconstriction of resistance and cutaneous vessels[71,79].The actions of SNS are dominantly mediated by secreted neurotransmitters such as norepinephrine(NE)that is released by sympathetic nerve terminals and,to a lesser degree,by the adrenal medulla and by epinephrine(EPI)that is chiefly released into peripheral circulation by the adrenal medulla.
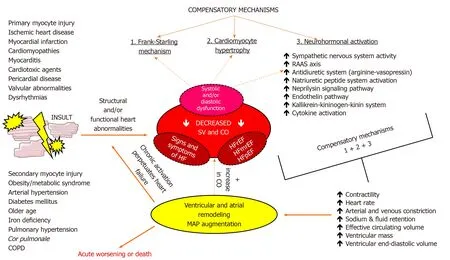
Figure 1 A diagram showing basic etiology,pathophysiology and compensatory mechanisms that are activated in heart failure.CO:Cardiac output;COPD:Chronic obstructive pulmonary disease;HF:Heart failure;HFmrEF:Heart failure with midrange ejection fraction;HFpEF:Heart failure with preserved ejection fraction;HFrEF:Heart failure with reduced ejection fraction;MAP:Mean arterial pressure;RAAS:Renin-angiotensin-aldosterone system;SV:Stroke volume.
Peripheral target organs are regulated by the two major sets of neurons serially connected to control the motor outflow of the SNS:(1)Preganglionic neurons that originate in the brainstem or the spinal cord;and(2)Postganglionic neurons that are part of sympathetic ganglia that are located outside of the central nervous system(CNS).Intrathoracic and extracardiac ganglia including stellate ganglia,middle cervical ganglia,and T2-T4 thoracic ganglia modulate the sympathetic outflow to the heart while sympathetic afferent impulses are carried through the dorsal root ganglia and reach the spinal cord,brain stem,and higher CNS centers.Cardiac sympathetic nerve fibers innervate myocardium at the subepicardial level,follow the path of major coronary arteries and are a predominant autonomic component in the ventricular tissue while parasympathetic nerve fibers,along with vagus nerve,run through subendocardium crossing the atrioventricular groove and are significantly more abundant in the atrial than ventricular myocardium thus exerting negative chronotropic effect with minimal effects on cardiac contractility[79].Furthermore,sympathetic innervation has a relatively higher density in the anatomical areas around sinoatrial node and coronary sinus while its density gradually increases from the base of the ventricle to the apex(positive base-to-apex gradient)[80,81].Intrinsic cardiac ganglia are located epicardially and receive innervation from post-ganglionic sympathetic and pre-ganglionic parasympathetic connections while most of sympathetic efferent and parasympathetic preganglionic fibers exhibit a large degree of intermixing thus most of the nerves reaching the heart in the mediastinum have mixed fibers(both sympathetic and parasympathetic components)[82].
The degree of SNS activation and sympathetic outflow to the heart and peripheral circulation,under physiological conditions,is regulated by a complex integration of autonomic cardiovascular reflexes.These reflexes include arterial baroreflexes,cardiopulmonary mechanosensitive reflexes,cardiac chemoreflexes,peripheral and central chemoreceptor reflexes,pulmonary stretch reflexes,cardio-cardiac reflexes and reflexes that are afferently projected from skeletal muscles[72].All of these reflexes have a common role in fine-tuning and maintaining adequate heart rate,mean arterial blood pressure,vascular tone,ventilation,and respiratory drive in response to various hemodynamic changes[83].These reflexes are listed with a summary of their function and potential impairment in HF(Table 1).According to modern pathophysiological findings,any depression of ventricular systolic function(irrespective of the underlying etiology)is augmenting cardiac reflex sympathoexcitation in chronic HF but might also be a leading culprit for the acute HF onset[72].For example,a physiological response to the sudden increase in the cardiac filling pressures should act to vasodilate venous capacitance vessels to accommodate for excessive fluid,however,paradoxical sympathetic discharge in HF instead causes vasoconstriction of venous pool(mainly splanchnic circulation)and redistributes fluid to cardiopulmonary pool thus precipitating congestion and causing dyspnea.For this reason,it could be that rapid increase in the effective circulating volume from the mobilization of fluid from the splanchnic bed is the dominant driving force behind increased central venous pressure and congestion encountered during HF decompensation episode and might depend on an external fluid gain to a lesser degree[84,85].Finally,cardiovascular-low threshold polymodal receptors are sensory endings localized in all cardiac chambers and large thoracic vessels that detect both mechanical and chemical stimuli and act in positivefeedback fashion with stimulatory effects on SNS[86].
In terms of prinicipal neurotransmitters that propagate the effects of SNS,NE is ejected in the synaptic cleft upon the stimulation from stellate ganglionsviapostganglionic fibers thus activating adrenergic receptors(ARs)in the heart and physiologically augmenting contractile strength,chronotropy,dromotropy and increasing mean arterial perfusion pressure.About 80% to 90% of released NE is reuptaken by the noradrenaline transporter 1 which is a monoamine transporter that clears NE from sympathetic nerve terminals/chromaffin cells while about 10% to 20%of remaining NE content is spilled into circulation[87,88].This NE turnover and metabolism can be evaluated with imaging methods such as scintigraphy by using radiolabelled guanethidine analogs of NE[89,90].Similarly,sympathetic fibers that innervate the adrenal gland stimulate chromaffin cells in the adrenal medulla that act as modified post-ganglionic fibers to release catecholamines in response to stressors or exercise.This efflux of catecholamines from adrenal medulla is predominantly comprised of EPI(about 80%)while NE makes up the remaining 20% with small amounts of dopamine being released into peripheral circulation as well[91].EPI and NE bind to specific ARs that are proteins embedded within the cell membrane with 7 transmembrane structures coupled to heterotrimeric G proteins.A total of two classes of ARs(alpha-and beta-adrenergic receptors)with 9 subtypes have been identified thus far:three α1receptors,three α2receptors and three β receptors(β1,β2,and β3)[92].A healthy human heart mostly consists of β1(75%-80%)and β2(20%-25%)adrenergic receptors and they represent the key effectors behind positive chronotropic and inotropic effects of catecholamines while β3adrenergic receptors(comprising less than 5% of total beta-receptor density)have been postulated to exert negative inotropic effects through upregulation of nitric oxide synthase pathway in human ventricle[93-95].It has been recently confirmed that β1and α1Breceptors are present in all ventricular cardiomyocytes[96].Alpha-1 adrenergic receptors(α1)and alpha-2 adrenergic receptors(α2)are chiefly expressed in vascular smooth muscle cells proximal and distal to sympathetic nerve terminals,respectively,and their activation elicits vasoconstriction of peripheral arteriolar and venous vessels while in the brain stem they modulate sympathetic outflow[97].A recent study by Beckeret al[98]showed that activation of neuronal endothelin B receptors can increase arterial blood pressure mediated through α1-adrenergic receptor signaling showing that abnormalities of endothelin system have a cross-talk with adrenergic systems in hypertension and HF[98].
Beta-adrenergic receptors act as powerful regulators of cardiac output and upon acute stimulation by catecholamines they facilitate fight-or-flight response while their chronic stimulation results in maladaptive and pathologic cardiac remodeling[99-101].Activation of β adrenergic receptors induces the activation of the stimulatory G protein(Gs)which further activates adenylyl cyclase leading to an increase in levels of intracellular cyclic adenosine monophosphate and activation of protein kinase A that phosphorylates several target proteins within the cardiomyocyte such as phospholamban,L-type calcium channels,troponin I,contractile proteins,and the cardiac ryanodine receptor and this mainly is the mechanism by which β1receptors regulate cardiac contractility/relaxation and heart rate[99,102].Furthermore,activation of β1receptors results in apoptotic and maladaptive remodeling signaling in the heartviaprotein kinase A-independent pathway mediated by Ca2+/calmodulin-dependent protein kinase II[99].On the other hand,β2receptors are distributed widely in the lungs,kidneys and blood vessels and possess a distinct function from the β1subtype as they are coupled both to Gsand inhibitory G protein(Gi)in cardiomyocytes and their activation enhances cardiac function and myocyte viability[103].
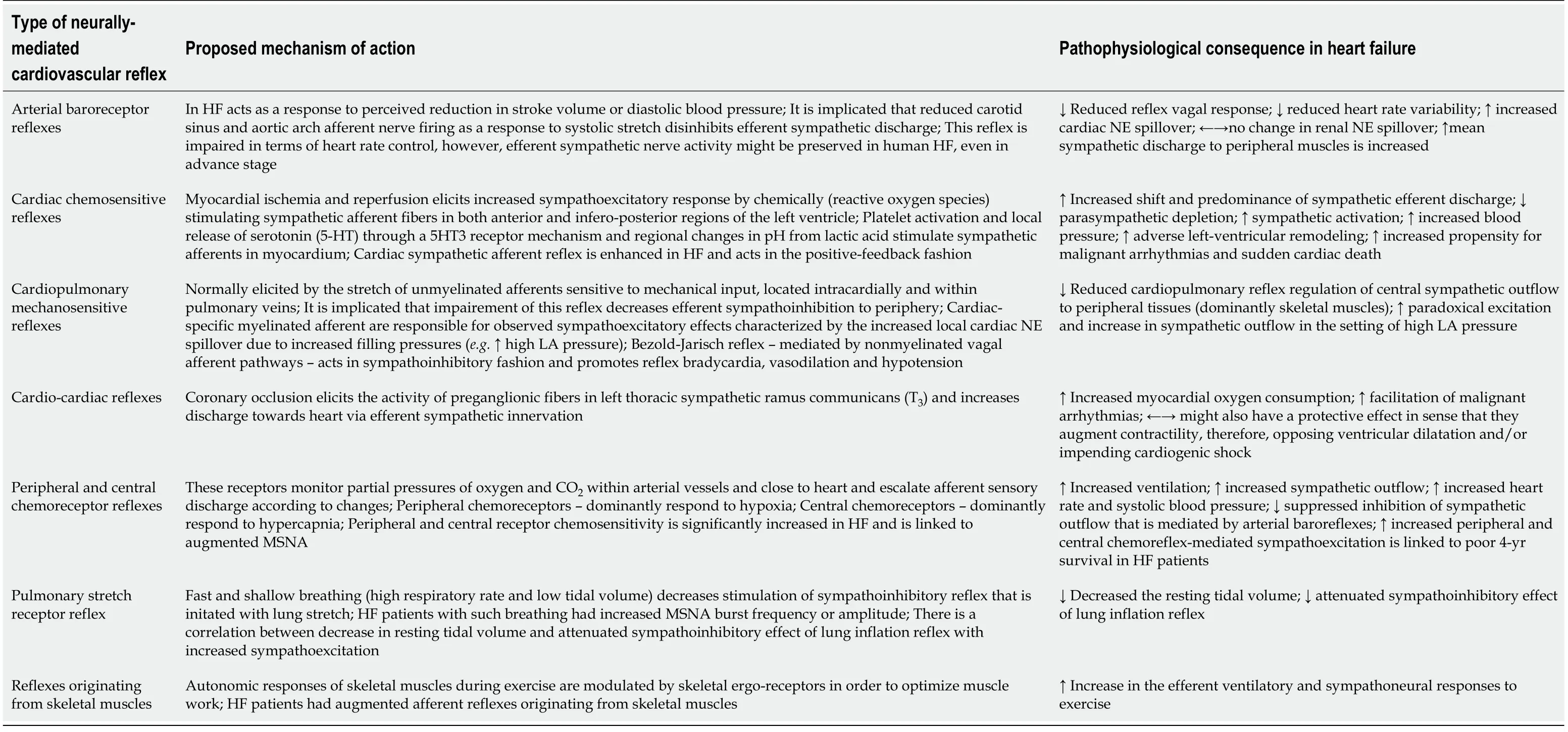
Table 1 Cardiovascular reflexes and their pathophysiological implications in heart failure
Finally,β3adrenergic receptors have a relatively minimal expression in the heart and they mediate unique downstream cellular effects once activated by catecholamines as they are mostly expressed in white adipose tissue where they mobilize stored fatty acids and regulate the release of adipokines while in brown adipose tissue they stimulate adaptive nonshivering thermogenesis[104].A study by Nappet al[105]showed that β3adrenergic receptors were mostly expressed in the endothelium of failing myocardium thus negative inotropic effect was most likely elicited by the NO liberation from the cardiac endothelial cells while β3stimulation itself seemed to deactivate rather than activate endothelial NOS[105].A study by Dessyet al[106]showed that β3receptors are abundantly expressed in the microvasculature of human coronary arteries in which their activation caused vasodilatation through NOdependent pathway and vessel hyperpolarization[106].Of note,the expression of β3adrenergic receptors in diverse cardiovascular pathologies seems to be upregulated and resistant to desensitization while in normal heart their activation resulted with a moderate negative inotropic effect[107,108].Similarly,in septic cardiomyopathy,functional β3receptors were upregulated and they increased negative inotropic response to β3agonists[109].Furthermore,in the setting of HF,activation of these receptors conferred beneficial effects with respect to excitation-contraction coupling and electrophysiological and mechanical remodeling of cardiomyocytes while also mediating vasodilatative pathways when β1and/or β2receptors are inoperative[110].In the clinical and translational realm,relevant studies confirmed these initial findings as they showed that the third-generation beta-blocker,nebivolol,exhibited agonistic action on β3adrenergic receptors in human ventricle thus providing evidence that highly selective blockade of β1receptors coupled with NO-dependent endothelial vasodilatation and neoangiogenesis in coronary microcirculation could improve cardiac energetics[111,112].Taken together,in the HF context,β3adrenergic stimulation might confer cardioprotection by attenuating excessive catecholaminergic stimulation mediated by β1adrenoceptors thereby presenting an attractive therapeutic target.The physiological effects of beta-adrenergic receptors are summarized and shown in Figure 2.
Finally,the expression of β-adrenergic receptors is physiologically modulated through G protein-coupled receptor kinases(GRKs),β-arrestins and complex intracellular signalosome[113].GRK family consists of seven different protein kinases that canonically recognize and phosphorylate agonist-activated G protein-coupled receptor signaling and initiate downstream β-arrestin-mediated cellular pathways[114].β-arrestins have a crucial role in the desensitization of activated seven transmembrane receptors such as β-adrenergic receptors,and they are key mediators of receptor endocytosis,ubiquitylation,and G-protein-independent cellular signaling[115].Therefore,it becomes obvious that the normal expression of GRKs is a cellular prerequisite to maintain physiological homeostasis regarding β-adrenergic receptor turnover by phosphorylation,degradation or clathrin-mediated receptor downregulation and internalization[116].
SYMPATHETIC NERVOUS SYSTEM PATHOPHYSIOLOGY AND ADRENERGIC DYSREGULATION IN HEART FAILURE
A chronic SNS overactivity is one of the key pathophysiological mechanisms that are operative in HF.In the acute phase,this upregulated SNS activity is an essential compensatory response initiated in order to counteract reduced contractility,however,in the long-term,it becomes a major contributor to cardiac dysfunction as it promotes maladaptive cardiac hypertrophy and cell death.
In a seminal study performed more than three decades ago,Swedberget al[117]showed that patients with chronic HF(CHF)had significantly higher arterial and coronary sinus venous NE concentrations compared to patients without HF while the net myocardial NE release in patients with CHF was about 20 times higher than that in patients without CHF[117].This was subsequently confirmed by Viqueratet al[118]demonstrating that endogenous plasma levels of NE and dopamine were significantly higher among patients with CHF compared to patients without CHF thus reflecting enhanced sympathetic activity in response to failing heart[118].Such overt sympathetic activity in HF closely paralleled increases in pulmonary artery pressures while activation of noradrenergic neurons in the brain might also be an underlying CNS mechanism of generalized sympathoexcitatory response observed in HF[119,120].In fact,it has been shown that the RAAS axis is the major regulator of the SNS activity in the brainviaangiotensin II type 1 receptors[121].This likely occurs due to the upregulated expression of angiotensin II type 1 receptors(promoting sympathoexcitation)and decreased expression of angiotensin II type 2 receptors(promoting sympathoinhibition)in the rostral ventrolateral medulla[122].Likewise,historical studies showed that 24-h urinary excretion of NE,EPI,and their O-methylated metabolites – normetanephrine and metanephrine was significantly higher in patients with congestive HF and reflected functional disease severity as assessed by the New York Heart Association(NYHA)class[123,124].Furthermore,there is not only a significant increase in circulating catecholamines but there is also an augmented neuronal NE spillover due to increased cardiac sympathetic nerve activity(SNA)while renal SNA nearly reached its maximum in the state of HF and showed to be an independent predictor of mortality in HF[125-127].Recent research efforts demonstrated that NE spillover does not only depend on increases in SNA but it also partially occurs due to mechanisms controlling NE release and reuptake in the synapse and these mechanisms seem to be deranged in HF[128].Increased NE spillover is in most cases paralleled by the reduced neuronal NE reuptake thus higher net concentrations of NE are present in the sympathetic synaptic cleft which further desensitizes myocardial βadrenergic receptors[129,130].A study by Haskinget al[131]further showed that cardiac and renal NE spillover in subjects with congestive HF was increased by 540% and 206%,respectively,compared to patients without HF while adrenomedullary-mediated EPI spillover was also markedly increased among these patients[131].
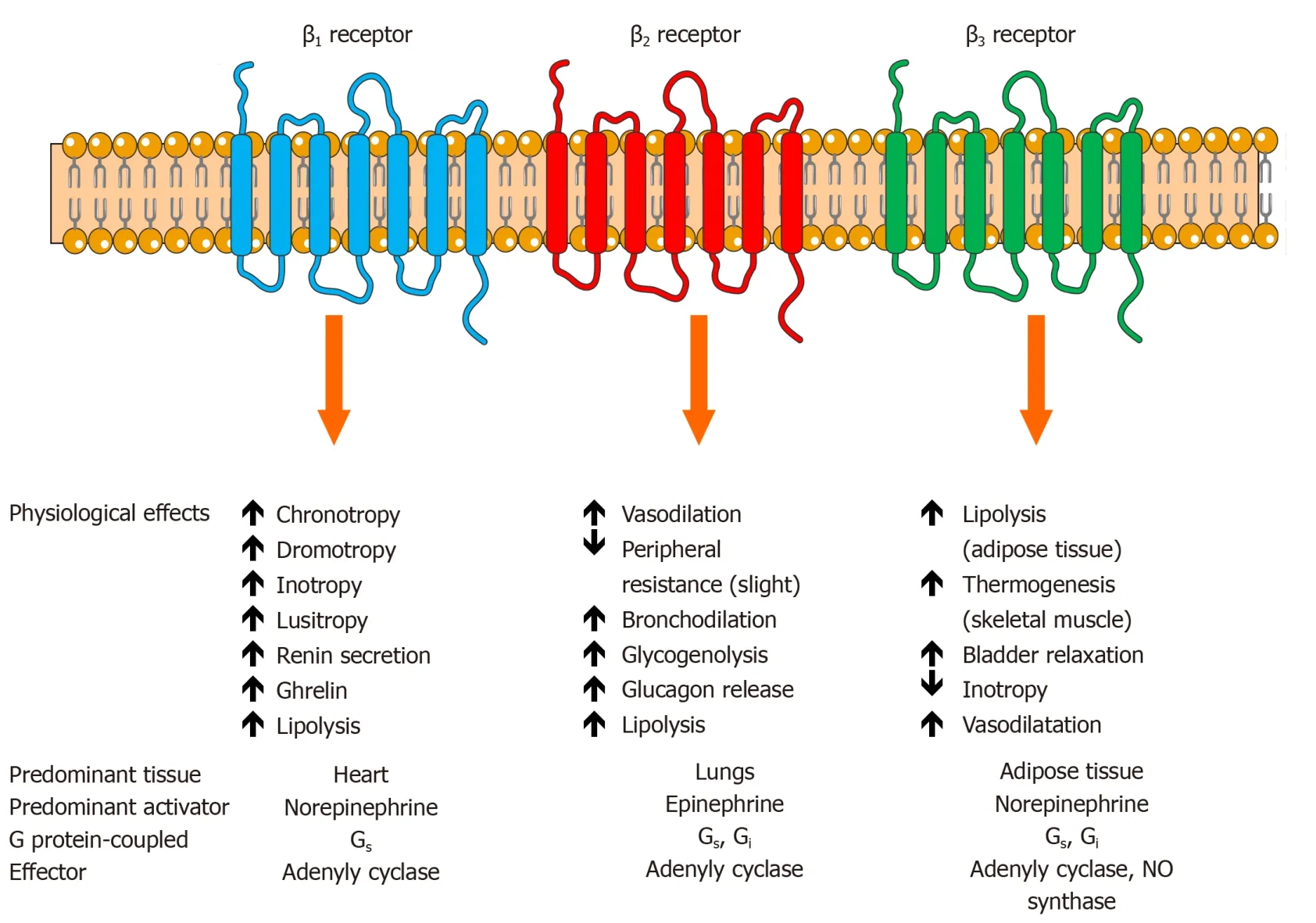
Figure 2 The function and physiological actions of beta-adrenergic receptors and adrenergic signaling.β:Beta;Gi:Inhibitory alpha subunit of G protein;Gs:Stimulatory alpha subunit of G protein;NO:Nitric oxide.
As previously mentioned,in a failing human heart,an important pathophysiological characteristic is a decreased sensitivity of β-adrenergic receptors to catecholamines while β-receptors are downregulated and decreased in their density and quantity[132].For example,β1adrenergic receptors are reduced up to 50% in HF and there is a 200% increase in Gi-mediated cellular pathways with concomitant significant upregulation of GRK2 activity(also known as β-adrenergic receptor kinase 1 or βARK1)that further promotes adrenergic receptor internalization[133].Myocardial GRK2 activity and expression have been increased in the failing heart as shown in several studies[134].Conversely,experimental inhibition of βARK1 resulted in a marked reversal of ventricular dysfunction[135].Finally,a wide variability of HF phenotypes and different response to HF treatment might suggest variants and functional polymorphisms of beta and alpha-adrenergic receptor genes[79].Some pharmacogenomic studies suggested that polymorphisms in β1-adrenergic receptors might affect susceptibility to HF such as Gly389 allele and Gly389 homozygotes;improved response to β-blocker treatment among Arg389 homozygotes while none of the candidate polymorphisms was an independent predictor of prognosis in HF[136,137].Likewise,specific β2-adrenergic receptor polymorphisms were linked with lower myocardial infarction rate and improved reverse left ventricular remodeling among patients with HF[138,139].
From the structural perspective,catecholamine spillover is cardiotoxic and its overexpression promotes senescence and inflammation of cardiomyocytes,upregulates tumor suppressor p53 pathway,and production of adhesion molecules by endothelial cells and macrophages and mediates cardiac dysfunction[140].Chronic and persistent stimulation by catecholamines in HF causes interstitial fibrosis,myocyte hypertrophy,oxidative stress,and impairs the responsiveness and function of cardiac β-adrenergic receptors[141].Engelhardt and colleagues experimentally demonstrated that increased chronic stimulation of β1adrenergic signaling resulted in a significant cardiomyocyte hypertrophy and apoptosis resulting in a marked loss of contractility and progressive reduction of LVEF with histological and functional deficits typical of HF[142].Catecholamine toxicity and generalized autonomic storm also have an important pathophysiological role in causing acute stress-related cardiomyopathies such as Takotsubo cardiomyopathy,acute LV dysfunction associated with subarachnoid hemorrhage,pheochromocytoma,and exogenous catecholamine administration as well as acute LV dysfunction in critically ill[143].Contrary to this,activation of β2adrenergic receptors delivered an antiapoptotic signal to cardiac myocytes through Gi-dependent coupling to phosphoinositol 3-kinase[144].Furthermore,it seems that the number of β2-adrenergic receptors does not change significantly in HF[145].These findings suggest that a fine balance between proapoptotic and antiapoptotic pathways initiated by differential adrenergic signaling is of fundamental importance for physiological cardiomyocyte function[146].
Importantly,studies have shown that the activation of SNS in the course of heart failure exhibits specific temporal dynamics and regional sympathetic profile.Rundqvistet al[147]showed that a selective increase in cardiac NE spillover(defined as increased amounts of NE at neuroeffector junctions)in patients with mild-to-moderate CHF was higher for more than a three-fold compared to healthy subjects while total body and renal NE spillover,as well as sympathetic outflow to skeletal muscles,were not different in HF patients compared to healthy controls[147].This study clearly showed that in the early stages of HF,selective increase in cardiac adrenergic drive precedes generalized sympathetic hyperactivity and outflow towards the periphery(skeletal muscles and kidneys)which is characteristic of advanced HF.In the early stages of HF,such cardiac sympathoexcitation might trigger ventricular arrhythmias and is associated with poor prognosis[126,148].Furthermore,local cardiac NE spillover might be the first component required for further β-receptor downregulation and depletion,adverse myocardial remodeling,depletion of NE stores,and impairment in G-protein signaling pathways,as discussed earlier.This might further drive hemodynamic deterioration and progressive LV dysfunction.Even more,blunted response and withdrawal of parasympathetic cardiac control seem to precede sympathetic activation during the development of HF.In support of this claim,in the tachycardia-induced model of HF,Ishiseet al[149]showed that parasympathetic withdrawal occurs rapidly and correlates with the decline in LV contractility and plasma NE increased gradually as LV diastolic function worsened while all of these changes recovered toward baseline values once pacing was ceased[149].The proposed mechanism was that depressed contractility resulted in the attenuated stimulation to the carotid sinus baroreceptor which diminished vagal efferent activity towards the heart thus demonstrating parasympathetic tonic withdrawal.Together,these findings suggest that in the course of SNS dysfunction in HF,sympathovagal imbalance might occur earliest as evidenced in parasympathetic withdrawal while sympathetic hyperactivity likely first occurs at the cardiac level before it is propagated to peripheral tissues and organs as observed in the advanced stages in HF.
Furthermore,dysfunction of cardiac reflexes is a hallmark of SNS hyperactivity in HF and it occurs to a similar degree regardless of HF etiology(ischemic or nonischemic)[150,151].There is a diminished baroreflex sensitivity in HF characterized by the marked suppression of inhibitory SNS reflexes such as arterial baroreceptor reflex while excitatory SNS reflexes such as those fired from peripheral chemoreceptors are enhanced[152].Floraset al[153]showed that a failing heart reacts to increased cardiopulmonary filling pressures through responsive and sensitive arterial baroreflex that elicits potent sympathoexcitatory hemodynamic actions[153].Furthermore,even among patients with mild CHF,an SNS-inhibiting baroreceptor function is already significantly impaired thus implying that baroreflex dysfunction might be one of the earliest constitutive phases in SNS activation during the natural course of CHF[154].Reduction in baroreflex sensitivity is even more severe if obesity and arterial hypertension are present among HF patients[155].Conversely,baroreflex activation therapy in HF,encompassing the deployment of a device electrically stimulating carotid sinus,succeeded in improving muscle sympathetic nervous activity and relevant clinical indices thus showing that modulation of autonomic balance in HF might improve relevant outcomes[156,157].
Collectively,these findings are of clinical relevance because ANS imbalance and predominance of sympathetic excitation cause electrophysiological perturbations in the vulnerable cardiac syncytium and can initiate arrhythmogenesis[158].For example,simultaneous stimulation of both sympathetic and parasympathetic systems can trigger AF while increased sympathetic stimulation is a contributing culprit to initiation of ventricular fibrillation(VF)or ventricular tachycardias(VT)or sudden cardiac death(SCD)[159].Beat-to-beat variability of ventricular action potential duration is increased with elevated sympathetic activity in HF patients and might precipitate ventricular arrhythmias while beta-blocker,bisoprolol,attenuated these effects[160].It was previously shown by Brunner-La Roccaet al[161]that high cardiac sympathetic activity in HF was an independent risk factor for sudden death,especially if sympathetic innervation was intact[161].Sympathetic denervation in the heart combined with the presence of high NE levels is tightly correlated to progression of HF and SCD[162].From the other way around,stellate ganglion blockade was effective in the acute reduction of ventricular arrhythmia burden and suppression of electrical storm thus clinically validating the concept that attenuation of sympathetic outflow to the heart from sympathetic ganglia can indeed mitigate the risk of future arrhythmic events[163-165].These clinical observations were inspired by the previous animal study demonstrating that spontaneous high-amplitude discharge activity from left stellate ganglion was strongly associated with the induction of malignant ventricular arrhythmias[166].Modern state-of-the-art neuromodulation strategies that are capable of mitigating VT/VF and atrial arrhythmias are,therefore,focused on increasing parasympathetic drive and inhibiting sympathetic neurotransmission[167,168].
Finally,it should also be noted that the widespread SNS activation also affects the function of skeletal muscles and promotes exercise intolerance in HF.Of note,diminished exercise capacity in terms of reduced peak oxygen uptake is present among HF subjects and is related to increased efferent sympathetic traffic to skeletal muscles,compared to control subjects[169].This study also showed that resting muscle SNA is inversely related to peak oxygen uptake thus suggesting that there is a peripheral neurogenic limit to exercise in HF.As later validated,this reduced exercise capacity in HF is more dependent on sympathetic outflow to skeletal muscles than to cardiac sympathetic outflow,as assessed by NE spillover[170].Furthermore,a subsequent study showed that muscle SNA was significantly higher while peak oxygen uptake was significantly lower in patients with ischemicvsnonischemic cardiomyopathy[171].The most recent clinical study also demonstrated that the αadrenergic-mediated vasoconstriction in HFrEF patients elicited a marked decrease in exercising skeletal muscle blood flow thus contributing to reduced exercise capacity in this population[172].Finally,HF patients present with a high degree of chronotropic incompetence and attenuated heart rate response to exercise which is partially due to postsynaptic desensitization of the β-adrenergic receptor pathways[173].
CARDIAC IMAGING AND SYMPATHETIC ACTIVATION IN HEART FAILURE
A noninvasivein vivoimaging modalities can assess sympathetic innervation of the heart and for these purposes single-photon emission computed tomography and positron emission tomography(PET)are used by employing radiolabeled analogs of NE.The myocardial uptake of these radioanalogs dominantly represents presynaptic nerve function and their density in the heart.The most commonly used single-photon emission computed tomography tracer is123I-metaiodobenzylguanadine(123I-mIBG)while most common PET tracer in clinical use is11C-hydroxyephedrine(11C-HED)[174,175].
Recent studies demonstrated that impaired myocardial sympathetic innervation and regional sympathetic denervation,as detected by the presence of11C-HED by PET imaging,were independently associated with grade 2-3 diastolic dysfunction and contractile dysfunction and fibrotic burden among patients with HFpEF,respectively[176,177].Similarly,data from prospective HF cohort studies demonstrated that diminished123I-mIBG uptake quantified as the reduced heart-to-mediastinum uptake ratio(H/M,indicating neuronal function including uptake and release of123ImIBG)or increased myocardial123I-mIBG washout rate(indicating higher adrenergic drive)were strong markers of abnormal myocardial sympathetic innervation and consistent predictors of poor prognosis among patients with HF[90,178-180].Furthermore,the ADMIRE-HFX study confirmed that H/M remained as a significant and independent predictor of all-cause mortality and the composite endpoint of death or death-equivalent events among nearly thousand NYHA II-III HF subjects during the median of 24 mo follow-up[181].
An elegant study by Wakabayashiet al[182]exploring123I-mIBG kinetics in terms of underlying HF etiology showed that123I-mIBG activity provided independent longterm prognostic information for both ischemic and non-ischemic etiologies of HF with lower H/M values having a greater impact on cardiac death among patients with ischemic compared to non-ischemic cardiomyopathy[182].In concordance with such findings among HF patients with ischemic cardiomyopathy,11C-HED PET-based studies revealed that regional myocardial sympathetic denervation and volume of denervated myocardium accurately predicted the risk of sudden cardiac arrest thus clearly correlating SNS innervation abnormalities with future arrhythmogenic events[183,184].Similar findings were confirmed by another research group showing that denervated myocardium quantified using PET strongly predicted the risk of sudden cardiac arrest,independent of LVEF,infarct volume and other clinical variables among HF patients with ischemic cardiomyopathy and with LVEF <35% that were eligible for implantable cardioverter-defibrillator device for primary prevention[185].Finally,the most recent study conducted among patients admitted for acute decompensated heart failure and prospectively enrolled in the OPAR registry demonstrated that patients with cardiac sympathetic nerve dysfunction,defined as low late H/M,had a significantly greater risk of future adverse cardiac events,irrespective of clinical phenotype based on the LVEF values[186].This study also showed that even a mild impairment in cardiac contractility(as shown in borderline LVEF values represented in HFmrEF cohort)was associated with sympathetic nerve dysfunction and was independently linked to poor outcomes thus suggesting that use of beta-blocker therapy in patients with HFmrEF phenotype is a viable pharmacotherapeutic option,as also supported by expert consensus statement and data from a large metaanalysis[73,187].
Taken together,these studies suggest that non-invasive cardiac imaging with norepinephrine analogs provides a reliable estimation of cardiac sympathetic nerve activity and this activity is strongly associated with clinical outcomes,regardless of clinical phenotypes or if HF is of chronic or acute onset.Such findings validate the concept that SNS overactivity is an important pathophysiological target in HF that must be efficaciously treated to improve outcomes and prevent sudden cardiac death.
HEART RATE VARIABILITY
Heart rate variability(HRV)is an established and widely used noninvasive method for the assessment of autonomic modulation of heart rate.It uses electrocardiographic(ECG)signal to measure subtle variations in the beat-to-beat heart intervals and is considered as a surrogate parameter of the complex interaction between CNS and cardiovascular system[188,189].These periodic oscillations in heart rate signals are transformed into different frequency areas and their relative intensity is reported as a numerical value[190].Briefly,low-frequency power(LF)and high-frequency power(HF),as well as the LF/HF ratio,are the most commonly used parameters in HRV analysis[189,191].In most of the studies,HF power is regarded as a surrogate of PNS activity while LF power is modulated by both SNS and PNS.Likewise,high LF power values are associated with increased sympathetic activity while the LF/HF ratio reflects global sympathetic/vagal balance[191].Generally,decreased HRV is associated with various pathologies and decreased life expectancy in several studies[188].
Regarding cardiovascular diseases,depressed HRV has been associated with autonomic neuropathy,heart transplantation,congestive HF,MI,and other incident cardiac conditions[192,193].Most data for low HRV and increased mortality have been corroborated from studies investigating populations with cardiovascular diseases such as post-MI patients,patients with HF and those experiencing SCD,and in contrast to this,such associations of HRV were historically more diluted when it comes to risk stratification among the general asymptomatic population[194].However,a recent study by Hillebrandet al[195]showed that low HRV was associated with a 32%-45% increased risk of a first cardiovascular event in populations without known cardiovascular disease[195].In a similar fashion,abnormal HRV parameters were independently associated with incident CHF in asymptomatic older adults[196].
In the setting of a failing heart,HRV is significantly reduced in most patients and associated with the high risk of death due to progressive HF,SCD and syndrome severity[197-199].Ponikowskiet al[200,201]demonstrated that depressed HRV on 24-h ambulatory ECG monitoring was an independent risk factor for poor prognosis in patients with CHF and was related to a higher risk of ventricular tachycardia[200,201].Similar findings were also confirmed in patients hospitalized for decompensated HF[202].An important study by Poussetet al[203]showed that a beta-blocker,bisoprolol,administered in a single dose of 5 mg per day managed to reduce heart rate and significantly increase HRV as per 24-h Holter ECG monitoring among patients with HF[203].This effect was attributed to the increased parameters of parasympathetic activity in HF thus showing that increased vagal tone may be responsible for the protective effect of beta blockers and may provide prognostic implications in HF.Similarly,beta-blockers improved cardiac autonomic regulation during high sympathetic stress of decompensated HF[204].
However,the foundational framework that links low-frequency and high-frequency components of HRV with sympathetic and parasympathetic nervous system division was developed decades ago and this algorithm does not integrate findings and data on HRV that were gathered in the past 30 years thus might have certain limitations in clinical practice[205].Another potential limit for the use of HRV in risk stratification of HF patients might lie in the fact that these parameters tend to be very low in most HF subjects,therefore,data dispersion might be small thus limiting survival regression models while many confounding non-neural factors might affect HRV values in HF[206].The future of risk stratification of events in HF likely lies in the improvement of HRV spectral analyses algorithms and integration of HRV data with other biosignals acquired from novel HF devices,imaging methods,and laboratory biomarkers.
LABORATORY BIOMARKERS OF SYMPATHETIC NERVOUS SYSTEM ACTIVATION IN HEART FAILURE
Laboratory biomarkers that can be measured in the peripheral circulation of HF patients can give us insight on underlying pathophysiological mechanisms that are occurring in patients with both acute and chronic HF.Since HF is a complex syndrome characterized by the high prevalence of comorbidities an integrated approach using multiple biomarkers could aid in the diagnosis,accurate risk stratification regarding mortality and future hospitalizations and perhaps enable optimal tailoring of pharmacotherapeutic and/or device therapies for the individual HF patient[207,208].A wide array of novel biomarkers reflecting pathophysiological processes of myocardial stretch,matrix remodeling,myocyte injury,oxidative stress,inflammation,neurohumoral activation,and renal dysfunction are becoming increasingly studied and integrated into the process of care for HF patient and clinical decision-making[19].The early adoption of these novel biomarkers in modern clinical practice has a great potential to complement traditional biomarkers that are regularly used in the workup of HF patients such as N-terminal prohormone of brain natriuretic peptide(NTproBNP),brain natriuretic peptide(BNP),high sensitivity cardiac troponin(hs-cTn),soluble suppression of tumorigenicity 2 or C-reactive protein[19].
In this last section of the review,we will focus on both established and novel laboratory biomarkers that are implicated in the pathophysiology of SNS activation in HF and as such might be potentially used in clinical practice.The summary of pathophysiological effects,cellular mechanisms of action,circulating levels,and association of selected biomarkers with outcomes in HF is presented in Table 2.
Norepinephrine
As previously discussed,circulating plasma levels and urinary excretion of norepinephrine(NE)are significantly higher among patients with congestive HF compared to those without,reflecting elevated sympathetic drive[117,118,124].A recent study by Matsushitaet al[209]showed that the endogenous catecholamine surge might be the cause of urgently presenting acute HF by eliciting an abrupt and excessive rise in blood pressure leading to increased after-overload and volume-shift lung congestion[209].A few decades ago,Cohn and colleagues showed that plasma NE has been independently related to subsequent risk of mortality among patients with chronic congestive HF and was also higher among those that had progressive HF compared to patients that died suddenly[129].This was later confirmed in the V-HEFT II study that enrolled patients with congestive HF showing that plasma NE was an independent predictor of prognosis and plasma NE values >900 pg/mL were associated with significantly greater mortality risk compared to lower NE tertiles[210].In the longitudinal follow-up of patients with HF from the Val-HeFT trial,changes of BNP and NE from baseline to 4 and 12 mo post-discharge significantly correlated to changes in morbidity and mortality[211].However,the administration of neurohormonal antagonists such as ACE inhibitors and beta-blockers in HF patients had variable and heterogeneous effects on circulating NE levels and there was a significant incongruency of these levels with endpoints such as mortality and reverse ventricular remodeling in a handful of relevant trials[212].These data suggested that reducing NE levels might not be the appropriate goal of neurohumoral antagonists and that NE is not a feasible laboratory biomarker of choice when it comes to measuring response to HF-directed pharmacotherapy.Finally,the fact that circulating NE measurements require high-performance liquid chromatography is a significant limitation to its wide use in clinical practice and imposes several analytical challenges and physiological limitations thus making it likely impractical as a routine biomarker in HF[213].
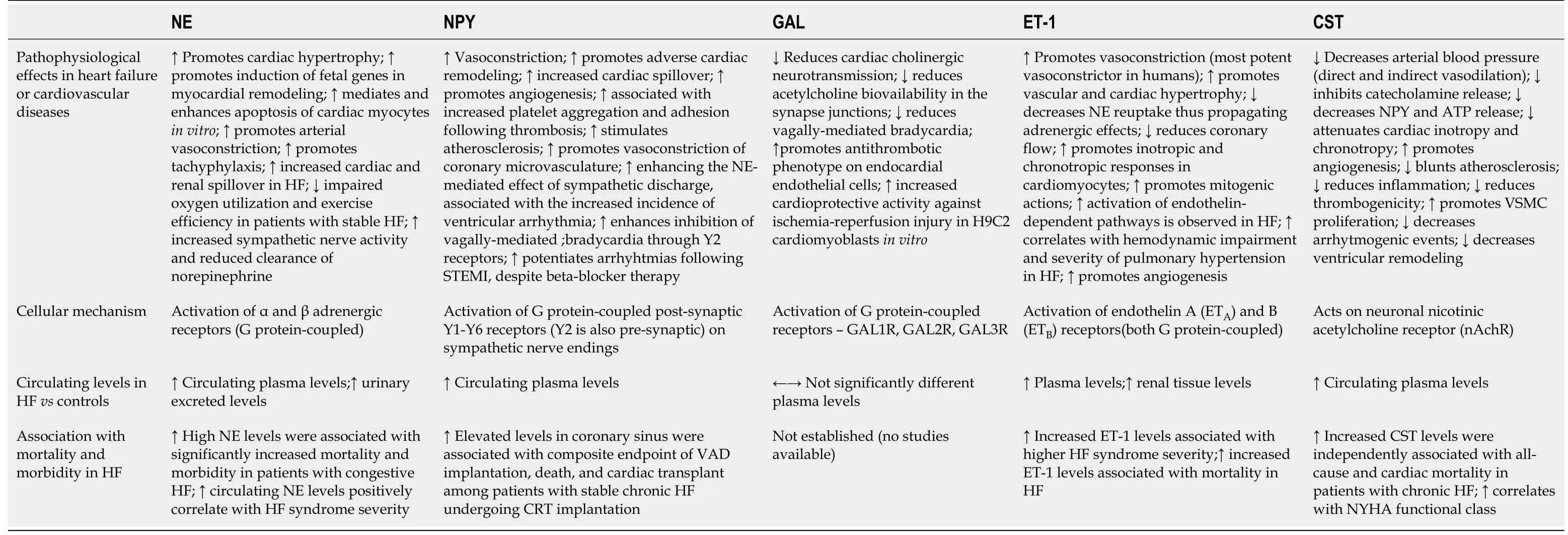
Table 2 Selected biomarkers in respect to their pathophysiological effects,cellular mechanisms,circulating levels and outcomes in heart failure
Neuropeptide Y
Neuropeptide Y(NPY)is a sympathetic co-transmitter with a longer half-life than NE and is widely distributed in the CNS and peripheral nervous system with pleiotropic physiological actions.In the cardiovascular system,NPY is co-released from cardiac sympathetic nerve terminals along with catecholamines(predominantly NE)and galanin[214].These sympathetic nerves supply vasculature,cardiomyocytes and endocardial endothelial cells in the ventricle while NPY physiologically modulates cardiovascular function,potentiates pressor effects of angiotensin II,elicits arterial and venous constriction,blunts parasympathetic activity,augments cardiomyocyte calcium loading,participates in cardiomyocyte remodeling and promotes angiogenesis[215-223].NPY and galanin have a direct ability to modulate vagus nerve to release acetylcholine and control heart rate while NPY plasma levels had a strong correlation with coronary microvascular function among patients with ST-elevation myocardial infarction[224].Maisel and colleagues were the first to report on elevated levels of plasma NPY in patients with congestive HF and this was later confirmed in several subsequent studies[225-227].
In the recent clinical study by Ajijolaet al[228],NPY was sampled from the coronary sinus(CS)among patients with stable CHF during the elective CRT device implantation[228].Researchers sought to answer if NPY as a peptide involved in adrenergic signaling is associated with outcomes among patients with stable CHF.They found that patients with NPY CS levels >130 pg/mL had significantly worse outcomes compared to those with lower NPY CS levels,even after adjusting for age,estimated glomerular filtration rate(eGFR),and LVEF(HR:9.5,95%CI:2.92-30.5,P<0.001)during the median follow-up of 28.8 mo while the composite endpoint consisted of death,ventricular assist device placement and cardiac transplant.Most of the signal from the composite endpoint was driven by death events and interestingly,CRT data at 6-mo follow-up showed that CS NPY levels did not significantly differ between CRT responders and non-responders(P= 0.76).Finally,immunohistochemical analyses revealed that sympathetic ganglia(stellate and middle cervical ganglion)of CHF patients contained less NPY compared to ganglia tissue obtained from healthy donors while no significant difference was observed in the NPY production between both groups as examined by the measured NPY mRNA levels.This study showed that CS NPY levels were elevated in stable CHF patients and associated with adverse outcomes and relevant clinical and laboratory characteristics while increased stellate ganglia sympathetic discharge was likely the culprit for these elevated levels.
Although CS NPY levels provided robust prognostic information among stable CHF patients,a problem in clinical practice arises in the peripheral venous sampling of NPY since those levels are not cardiac-specific and are mostly of hepatomesenteric origin since NPY has been identified as a stimulator to food intake[229].In cardiac failure,there is an increase in resting NPY spillover within the myocardium,however,the net overflow of NPY to plasma was dominantly from hepatic circulation,but not the cardiac,forearm or cerebral circulations showing a marked difference in regional distribution of NPY content[230].It has also been shown that sympathetic activation by exercise produced only a modest increase in cardiac NPY overflow without the concomitant change in arterial NPY concentrations finally concluding that plasma NPY concentrations are less sensitive than those of plasma NE in terms of quantifying SNS responses regulating the systemic circulation and cardiac hemodynamics in HF,as implied in some previous studies[225,230,231].Finally,a recent preclinical study showed that NPY blockade by experimental Nur77 agent protected against adverse cardiac remodeling by limiting NPY-mediated signaling(NPY-NPY1R)in the cardiomyocytes[232].Most important characteristics and effects of NPY are depicted in the Figure 3.In the future,antagonists of NPY receptors Y1 and Y2 might be a feasible therapeutic option in acute myocardial infarction but also during chronic HF and hypertension[224].These pharmacotherapeutic options would complement beta-blockers and implantable vagus nerve stimulators to improve outcomes in patients with cardiovascular diseases[224].
Galanin
Similarly to NE and NPY,galanin is an adrenergic co-transmitter with a short half-life(about 5 min)released from peripheral postganglionic neurons and is implicated in attenuation of cardiac cholinergic tonus after burst sympathetic activity thus contributing to autonomic imbalance and the pathophysiological phenomenon known as“sympathovagal crosstalk“[224,233].This phenomenon can remain chronically activated and sustained even in the presence of beta-adrenergic blockade thereby it could be a valid therapeutic target in the spectrum of neurohumoral activation in HF[224].Furthermore,nerve terminals of parasympathetic neurons in the heart express both galanin receptors and NPY receptors(NPY Y2)which,upon activation,reduce acetylcholine release[233].During the prolonged sympathetic activation there is a release of a slowly diffusing co-transmitter galanin,together with NPY,that bind to these receptors and reduce cholinergic neurotransmission in the heart[234].Furthermore,galanin through its receptors interacts with other neuropeptides such as NPY and angiotensin II and their receptors,namely Y1and AT1thus having a potential role in neurochemical modulation of central cardiovascular control[235].
A recent prospective case-control study in the clinical realm showed that unlike pro-BNP,copeptin and NPY,galanin levels were similar among patients with HF patients and control subjects while pro-BNP was the only significant determinant of galanin levels in HF patients[236].Authors postulated that galanin most likely has a predominant paracrine modulatory function at the level of peripheral cardiac sympathetic nerves,therefore,its circulating levels in plasma might not reflect the degree of its local involvement in sympathovagal crosstalk.Finally,since natriuretic peptides promote catecholamine release from cardiac sympathetic neurons,authors suggested biological plausibility of their finding that galanin positively correlated with BNP[237].On the other hand,galanin promoted anti-thrombotic phenotype on cultured endocardial endothelial cells from HF patients through attenuation of von Willebrand factor extrusion and multimer expression while this effect was not elicited by the NPY[238].One preclinical study in the animal model of HF showed that galanin receptor type 1 agonist improved cardiac function and attenuated ventricular remodeling[239].Most important characteristics and effects of galanin are depicted in Figure 3.Due to the scarcity of studies examining the role of galanin in HF,future preclinical and clinical studies are warranted to further elucidate its biological functions and its potential as a biomarker in HF.
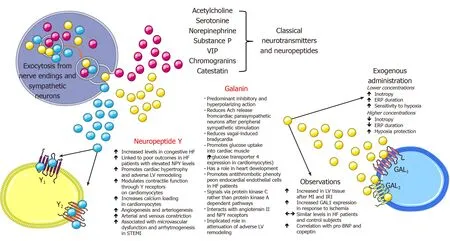
Figure 3 Physiological and pathophysiological implications of neuropeptide Y and galanin with respect to cardiovascular system.Ach:Acetylcholine;ERP:Effective refractory period;HF:Heart failure;IRI:Ischemia-reperfusion injury;LV:Left ventricular;MI:Myocardial infarction;NPY:Neuropeptide Y;STEMI:ST-elevation myocardial infarction;VIP:Vasoactive intestinal peptide.
Endothelin
Endothelins represent a family of three similar 21 amino acid length peptides –endothelin 1(ET-1),2(ET-2)and 3(ET-3)of which ET-1 and ET-2 bind to G-protein coupled endothelin receptors A(ETA)and B(ETB)on vascular smooth muscle cells with equal affinity to both while ET-3 exhibits lower affinity for ETArelative to ETBreceptor[240].Of all endothelins,ET-1 is predominantly produced by vascular tissue,has inotropic,chemotactic and mitogenic properties,induces collagen synthesis by cardiac fibroblasts,and is biologically the most potent vasoconstrictor in the human cardiovascular system[241].Furthemore,autocrine binding of ET-1 to ETBreceptors promotes NO and prostaglandin release and consequent relaxation of vascular smooth muscle cells.ET-1 plays a role in neuronal development,growth,and function while biologically promoting vascular and cardiac hypertrophy,inflammatory responses and is an independent factor contributing to exacerbation of the cardiovascular disease[242-244].The main source of ET-1 and its precursor,big endothelin-1(BigET-1)are pulmonary vascular endothelial cells,therefore,elevated plasma levels of ET-1 or bigET-1 might closely reflect the degree of pulmonary endothelial dysfunction in HF while ET-1 was significantly overexpressed in the lungs of patients with pulmonary hypertension[245-247].Stanglet al[246]demonstrated that in severe congestive HF lungs act as a producer while coronary and peripheral circulation act as consumers of BigET-1 and ET-1 while short-term vasodilator therapy decreased endothelins and restored pulmonary,coronary,and peripheral balance[246].Endothelin receptors are also expressed in the CNS and central administration of endothelin modulated endocrine and cardiovascular regulation,behavior and MAP[248].In the preclinical experiment,an injection of ET-1 in different regions of the brainstem of normotensive rats resulted in a differential response in heart rate,arterial blood pressure,and respiratory frequency indicating that endothelin has a modulatory role in cardiovascular function[249].
Previous studies showed that HF is associated with high levels of ET-1 in plasma and renal tissue and these levels correlated with syndrome severity,especially with the extent of pulmonary hypertension,and overall contributed to the progression of chronic HF[250-254].In a preclinical study,infusion of tezosentan(ET-1 antagonist)significantly decreased MAP in both normal and HF animals and reduced cardiac sympathetic nerve activity(CSNA)in normal animals,however,no decrease was observed in HF animals[255].Therefore,this study showed that endogenous levels of ET-1 contribute to the baseline levels of CSNA in healthy animals,however,this correlation was absent in experimentally induced HF.Contrary to this,a non-selective experimental ETAand ETBantagonist(TAK-044)suppressed sympathetic activity and improved arterial baroreflex function in rats with HF[256].Similarly,the addition of ACE inhibitor to ETAreceptor antagonist significantly improved cardiac failure after extensive MI in a rat model of congestive HF,compared with ACE inhibition monotherapy[257].A cross-talk between the endothelin system and the adrenergic system has been demonstrated as activation of ETBreceptors on sympathetic neurons caused an increase in arterial blood pressure through vasoconstriction mediated by α1-adrenergic receptors[98].Sympathoexcitatory effects are also promoted through the interaction of ET-1 with ETAreceptors as this resulted in cardiomyocyte hypertrophy through adrenergic signaling pathways and massive NE release while it also contributed to impaired responsiveness of renal mechanosensory nerves in congestive HF[258,259].In the rat model of HF,endogenous ET-1 impaired NE reuptake through activation of ETAreceptors while in a healthy heart ETA-mediated inhibition of NE reuptake was countered,but to a lesser degree,by the ETB-mediated silencing of NE release resulting in a net increase in left ventricular contractility suggesting that fine balance between NE reuptake and exocytotic release is modulated by endothelin signaling as it was also suggested in previous studies[260,261].
However,while endothelin pathway inhibition seemed promising in animal and preclinical models of HF,these observations did not translate to human clinical studies as ET-1 antagonist tezosentan did not improve symptoms or clinical outcomes in patients with acute HF although ET-1 levels were independently associated with short term in-hospital outcomes and 180-d mortality in patients hospitalized for acute HF,as demonstrated in ASCEND-HF substudy[262-264].A predictive value of BigET-1 in patients with left ventricular dysfunction after AMI on the composite endpoint of cardiovascular death or hospitalization for worsening HF has been demonstrated in the subanalysis from EPHESUS study,however,neurohumoral antagonist –eplerenone seemed to have no significant effect in modifying BigET-1 levels at followup[265].Authors proposed that levels of BigET-1(as a precursor of ET-1)likely reflect the degree of ET-1 synthesis while BigET-1 is also a more feasible laboratory biomarker due to its longer half-life than that of ET-1[266].This notion has been confirmed in a previous study that established how elevated plasma ET-1 levels in human CHF dominantly represent the elevation of Big-ET-1 while ET activity was not changed in CHF compared to a healthy state[267].Furthermore,increased ET-1 levels were detected only in moderate or severe CHF and not among asymptomatic patients or those with mild CHF while plasma concentrations in range 5-40 pmol/L seemed to exhibit vasoactive effects[267,268].Previous studies confirmed that BigET-1 provided prognostic information regarding the cardiovascular mortality during the 12-mo follow-up(HR:1.42,95%CI:1.04-1.95,P= 0.03),all-cause mortality during the 23-mo follow-up(HR:1.49,95%CI:1.20-1.84,P= 0.0003)and the composite endpoint of mortality and morbidity(HR:1.43,95%CI:1.20-1.69,P<0.001)at 23 mo,however,in the latter study BNP remained the strongest neurohormonal prognostic factor[269,270].In the small study that enrolled patients with severe CHF,Big-ET-1 and ET-1 levels were higher at baseline than in patients with mild to moderate CHF or healthy subjects and were found as robust independent predictors of survival,even beyond natriuretic peptide levels[271].
When 32 studies with 18497 HF patients were summarized in the meta-analysis,it was shown that plasma ET-1 and its related peptides were associated with poor prognosis and mortality in diverse spectrum of HF populations[272].On the other hand,a meta-analysis of randomized clinical trials showed that neurohumoral antagonism of ET receptors in HF patients improved cardiac output,pulmonary and systemic hemodynamics but had a modest effect on clinical outcomes[273].Therefore,these data suggest that there is a significant discrepancy between these observations – on one hand,ET signaling has been consistently associated with poor outcomes and prognosis in HF and on the other hand,pharmacological targeting of these adverse pathways seems less impressive in improving outcomes.
Perhaps there is a need to fine-tune and identify which subgroups of HF patients would have the greatest benefit from drugs interfering with ET pathways.In that regard,ET-1 and its fragments have shown some potential as valuable biomarkers among HFpEF patients with pulmonary hypertension or pulmonary dysfunction as its levels were associated with the degree of pulmonary hemodynamic derangements,reduced functional reserve of the right ventricle,diminished cardiac output and impaired cardiac response to exercise and peak oxygen consumption[274,275].Even in the general population,elevated plasma ET-1 levels were in strong relation with elevated pulmonary artery systolic pressures on the echocardiogram and correlated with mortality and incident HF[276].
Therefore,current data suggest that activation of the endothelin system may play an important role in the pathophysiology of pulmonary hypertension in HFpEF and that it might present a viable target and a step towards precision medicine approach in HFpEF[277].Regarding the potential ET pathway inhibition in HFpEF,thus far there are limited but encouraging preliminary reports.In the preclinical murine model of HFpEF,dual ETA/ETBblockade by macitentan improved HFpEF by abrogating aldosterone-induced cardiomyocyte hypertrophy and reducing stiffness through decreased expression of type I collagen and titin n2B in the left ventricle[278].In the clinical domain,in patients with HFpEF,ETAreceptor antagonist sitaxsentan improved exercise tolerance,however,failed to decrease left ventricular mass or improve diastolic function while the study was not powered for mortality and rehospitalization analyses[279].A small and prematurely stopped study showed that ET receptor blocker bosentan did not improve outcomes in HFpEF patients with pulmonary hypertension[280].Therefore,due to the size and scarcity of available studies,a question whether ET-1 antagonists would improve outcomes in HFpEF yet remains to be answered by future and adequately powered randomized controlled trials.It is possible that neurohumoral biomarkers such as endothelin and its derivatives will enable us a more precise phenotyping of HFpEF patients to identify those that have a significantly impaired pulmonary function and that would receive the greatest benefit from ET pathway-oriented therapeutic interventions.The summary of synthesis,cellular effects,and pathophysiological implications of ET-1 are presented in Figure 4.
Catestatin
Catestatin(CST)is a product of precursor hormone chromogranin A(ChgA)and was isolated in 1997 by Mahataet al[281].Its principal physiological action is the negative regulation of catecholamine release into circulation through the mechanism of noncompetitive and reversible antagonism of neuronal nicotinic cholinergic receptors(nAChR)[281,282].Its precursor molecule,ChgA,and other soluble secretory proteins are co-stored and co-released with catecholamines from vesicles in the neuroendocrine,endocrine and immune cells and sympathetic neurons thus have an important modulatory role of the adrenergic system[283].Upon stimulation of chromaffin cells or sympathetic axons,a marked elevation of ChgA levels was detected[284].Levels of ChgA are elevated in peripheral blood of patients with chronic HF and AMI and correlate with mortality and poor outcomes[285-287].Even more,an intramyocardial production of ChgA is established in humans and was associated with negative inotropic and lusitropic effects on the mammalian heart thus providing evidence for neuroendocrine regulation of cardiac function by ChgA[288].Furthermore,immunohistochemical biopsy studies showed that ChgA is co-localized with BNP in the dilated and hypertrophic left ventricle while ChgA levels correlated with enddiastolic left ventricular pressures[288].
CST is a 21 amino acid fragment derived from ChgA(ChgA352-372)and is secreted by neuroendocrine tissues and nerve endings while it is widely distributed in the secretory granules of skin,sensory organs,and myocardium[289].Its most important physiological effect is the autocrine action on the chromaffin cells in the adrenal medulla and adrenergic neurons by which it modulates spillover of catecholamines(primarily NE)into peripheral circulation while concomitantly exhibiting paracrine and endocrine effects since it can be readily measured in the venous and arterial blood[290].Furthermore,CST is potent regulator of arterial blood pressure since it exerts direct vasodilatative effect in humansin vivo,activates histamine release from mast cells and stimulates production of NO within endothelial cells[291-293].In the chromaffin cell,ACh is a physiological agonist that,upon activation of ionotropic nAChR receptor,permits Na+entry into the cell which further depolarizes cellular membrane and enables activation of voltage-gated Ca2+channels and subsequent Ca2+entry that mobilizes chromaffin granules and initiates exocytosis of several neurohormones,neuropeptides,and catecholamines[281,294].Once secreted outside of the cell through the process of exocytosis,extracellular post-translational proteolytic processing of the ChgA molecule will release several bioactive peptides and CST that will ultimately bind nAChR receptors of chromaffin cells in autocrine fashion thus antagonizing ACh actions in the periphery as depicted in Figure 5[295].
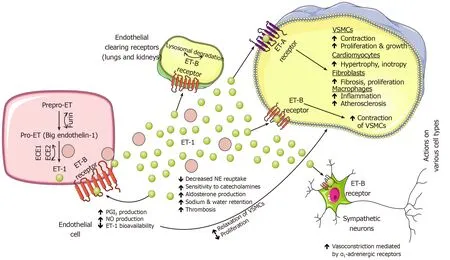
Figure 4 Physiological and pathophysiological implications of endothelin-1 in circulation and on various cell types and adrenergic neurons.ECE:Endothelin converting ezyme;ET-1:Endothelin-1;NE:Norepinephrine;NO:Nitric oxide;PGI2:Prostacyclin(prostaglandin I2);VSMCs:Vascular smooth muscle cells.
In the perspective of previously discussed catecholamine storage vesicle neurotransmitters,Mahapatra and colleagues demonstrated that CST inhibited nicotinically triggered exocytotic release of several co-transmitters from chromaffin granules such as NPY,adenosine triphosphate,chromogranins and catecholamines thereby demonstrating that CST is a potent regulator of neuropeptide transmission in the sympathochromaffin system[296].However,in the CNS,CST exhibits both sympathoexcitatory and procholinergic effects depending on the region of medulla where its expressed[297,298].Of established cardiovascular effects,CST suppresses betaadrenergic activation and acts in a negative inotropic and chronotropic fashion,stimulates angiogenesis and proliferation of vascular smooth muscle cells,decreases thrombogenicity of endothelial cells,suppresses atherosclerosis and inflammation while also exerts cardioprotective effects by abrogating cardiomyocyte ischemiareperfusion injury[299-306].A very recent study by Alamet al[307]showed that CST has a direct and independent inhibiting effect on hypertrophy elicited by NE in the cultured H9c2 cardiac myoblasts and that is involved in the regulation of a large number of fetal genes that are upregulated during the process of myocardial hypertrophy[307].Furthermore,the same study showed that CST effectively blunted stimulative effects of NE and other mitogenic signals on β1and β2adrenergic receptors thus providing novel evidence that CST has a direct modulatory effect on adrenergic transmission at the level of adrenergic receptors.Similarly,in the model of rat heart,CST activated β2and β3adrenergic receptors thus upregulating the activity of eNOS and consequently increasing cyclic GMP and phosphodiesterase type 2(PDE2)levels[308].
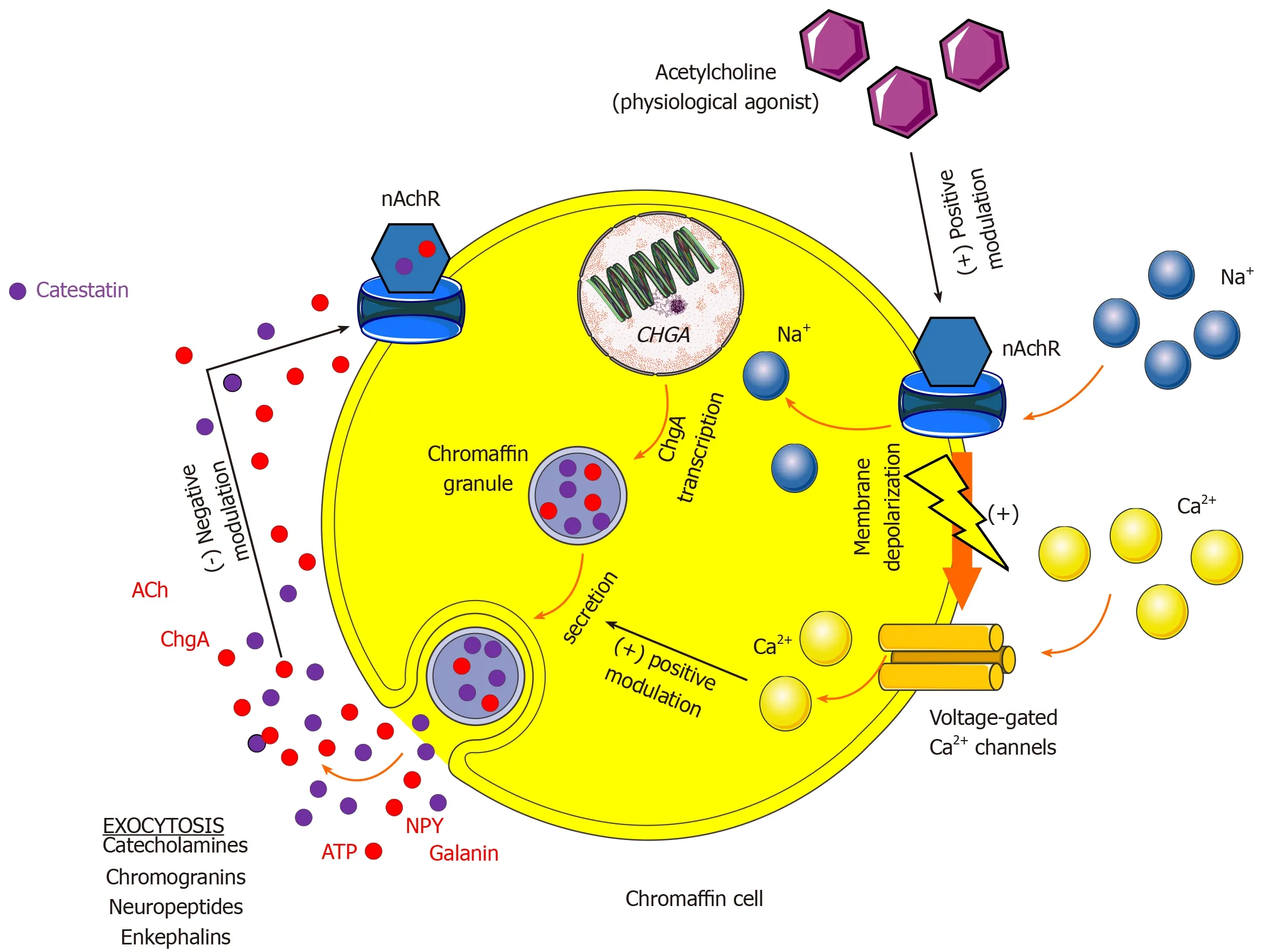
Figure 5 Mechanism of catestatin autocrine modulation of chromaffin cell in the adrenal medulla during sympathetic stimulation.ACh:Acetylcholine;ATP:Adenosine triphosphate;Ca2+:Calcium;ChgA:Chromogranin A;nAchR:Neuronal type of nicotinic cholinergic receptors;Na+:Sodium;NPY:Neuropeptide Y.
In line with its sympatholytic effects,chronic administration of exogenous CST improved autonomic function,shortened QT interval,and action potential duration and reduced the incidence of experimentally-induced ventricular arrhythmias in a rat model of myocardial infarction[309].Similarly,in the rat model of hyperadrenergic hypertension,rats with ablatedChgAgene showed significantly longer QT interval,Ramplitude,and QRS time-voltage and this was accompanied by increased resting heart rate and QT variability thus demonstrating that arrhythmogenic ventricular assault develops in the status of low circulating CST levels[310].These preclinical observations were clinically validated as elevated CST levels were an independent predictor of complicated malignant arrhythmias among AMI patients[311].This observation might seem counterintuitive at first,however,it could be explained in the sense that CST levels reflect a compensatory response for the increased SNS activity and excess catecholamine discharge and are attempting to restore autonomic balance.Therefore,circulating CST levels likely“mirror“ biological catecholamine turnover and degree of sympathetic activity as CST co-localizes and is co-released with catecholamines and other neuropeptides.Finally,cardioprotective effects of CST are likely pathophysiologically overpowered by sympathetic discharge in conditions in which cardiovascular homeostasis is disrupted such as AMI or decompensated HF,despite the relatively high circulating CST levels.
There are only a few available studies that examined the role of CST in HF and investigated its prognostic role in this syndrome.
In the first study by Zhuet al[312]CST levels gradually decreased from stage A to C of HF while there was a significant difference between stage A and B in terms of CST concentrations with a cut-off value of 19.73 ng/mL providing 90% sensitivity and 50.9% specificity for the detection of B stage of HF[312].This finding is of clinical relevance since stage B presumes structural cardiac disorder but without symptoms,while stage A assumes patients at high risk for developing HF but without functional or structural heart disorder.Therefore,this study showed that there is a utility for decreased CST levels implying structural heart disease among asymptomatic patients.In the study by Liuet al[313]performed in the similar setting,CST was found higher in patients with HF compared to control subjects and it positively correlated with functional syndrome burden as assessed by the NYHA class[313].Furthermore,etiology of HF(ischemic or not)and NYHA class predicted plasma CST levels while BNP provided better area under the curve value than CST in terms of detecting moderate to severe HF diagnosis(area under the curve values of 0.831 and 0.626,respectively).Adding CST to BNP did not improve diagnostic accuracy.
Recently,Borovacet al[314]showed in CATSTAT-HF study,that patients with acutely decompensated HF had higher serum CST levels if they belonged to a higher NYHA functional class while circulating CST levels were significantly higher among patients with ischemicvsnon-ischemic etiology of disease thus likely reflecting an augmented SNS neurohumoral activation in patients with ischemic etiology of the disease(in the study defined as those with the history of myocardial infarction)[314].The same study revealed that CST levels did not differ in respect to LVEF phenotypes while CST levels independently correlated with NYHA class,waist-to-hip ratio,HbA1c,LDL cholesterol,non-HDL cholesterol,high sensitivity cardiac troponin I and the heart rate at admission and rest.Finally,higher CST levels were strongly associated with favorable echocardiographic profile as they positively correlated with smaller LV volumes and dimensions,as well as with decreased left ventricular mass and smaller dimensions of the left atrium and this finding clinically validates the concept that CST locally has cardioprotective effects,attenuates adverse ventricular remodeling and acts in antihypertrophic fashion,as these biological effects were postulated in a few earlier preclinical studies[307,315].
CONCLUSION
Finally,the prognostic value of CST as a biomarker was demonstrated among chronic HF patients.In the multivariate Cox regression analysis,plasma CST was an independent risk factor for all-cause death(HR:1.84,95%CI:1.02-3.32,P= 0.042)and cardiac death(HR:2.41,95%CI:1.26-4.62,P= 0.008),respectively,during the median follow-up of 52.5 mo[316].If patients had both high CST and BNP levels during hospitalization the risk of all-cause death increased 3-fold while the risk of cardiac death increased 4-fold.
Based on these findings it is plausible that CST could be a reliable indirect marker of SNS activity and it is likely that high CST levels reflect advanced disease burden and high sympathoexcitatory profile of an individual HF patient.Furthermore,CST provides complementary prognostic information to natriuretic peptides in terms of mortality in HF and could aid in the risk stratification of chronic HF patients.However,since the latter finding is based on only one clinical study further large-scale studies are required to validate these findings and clarify the role of circulating CST levels in predicting HF prognosis.Finally,patients with elevated CST levels might be suitable candidates for the introduction or up-titration of sympatholytic agents such as beta-adrenergic blockers,however,the effects of neurohumoral antagonists on circulating CST levels are yet to be determined.
ACKNOWLEDGEMENTS
Figures presented in this manuscript are of author’s own design,however,they were constructed by using some of the illustration elements kindly provided by Servier.Servier Medical Art is licensed under a Creative Commons Attribution 3.0 Unported Licence.
 World Journal of Cardiology2020年8期
World Journal of Cardiology2020年8期
- World Journal of Cardiology的其它文章
- Oliver Wendell Holmes’1836 doctorate dissertation and his journey in medicine
- Systematic review and meta-analysis of outcomes of anatomic repair in congenitally corrected transposition of great arteries
- Impact of cardiologist intervention on guideline-directed use of statin therapy
- Forensic interrogation of diabetic endothelitis in cardiovascular diseases and clinical translation in heart failure
- Classic Ehlers-Danlos syndrome and cardiac transplantation-Is there a connection?