
胰岛β细胞保护策略研究进展
2020-10-29 09:26:02刘红霞蔡东成何朝勇
中国药科大学学报 2020年5期
王 静,刘红霞,蔡东成,何朝勇
(中国药科大学药学院药理系,南京211198)
糖尿病是以血液中葡萄糖水平升高为主要特征的代谢性疾病。随着生活水平不断提高,糖尿病发病率急剧上升。流行病学数据表明,2017年至2045年,糖尿病患者增长率将超过50%,糖尿病患者数预计达到6.93亿[1]。
胰岛素抵抗导致胰岛β细胞代偿性分泌更多胰岛素以维持机体正常的血糖水平,随病程延长,胰岛β细胞功能和数量逐渐下降[2]。早期胰岛β细胞死亡的可能机制包括线粒体功能障碍、氧化应激、内质网应激和糖脂毒性等。因此,恢复胰岛β细胞功能及体量对治疗糖尿病具有重要意义。本文基于糖尿病发病机制及胰岛β细胞死亡的不同作用机制综述了胰岛β细胞保护的最新研究进展,旨在探讨恢复胰岛β细胞功能及体量在糖尿病治疗中的必要性。
1 增强胰岛β细胞功能,促进胰岛素合成与分泌
胰岛β细胞是机体唯一可以合成并分泌胰岛素的细胞。促进胰岛素合成与分泌是增强胰岛β细胞功能的主要方式。
1.1 调节胰岛素合成相关靶点
胃饥饿素(ghrelin)是生长激素促分泌素受体的内源性配体,它可以促进生长激素的分泌,并与能量代谢息息相关。Gray等[3]发现,ghrelin显著提高大鼠前胰岛素原mRNA的表达,从而促进胰岛素的合成。同时,一些转录因子参与调控胰岛β细胞合成与分泌胰岛素。Yang等[4]发现,miR-96抑制转录因子Sox6活性增强胰岛素的合成与分泌,促进胰岛β细胞的功能。Villamayor等[5]报道在胰腺特异性敲除GATA结合蛋白6(GATA binding protein 6,GATA6)小 鼠 中 ,转 录 因 子 PDX1、NKX6.1和MafA等蛋白表达显著下调,未成熟的胰岛素颗粒增多,胰岛素合成减少,分泌受损。
1.2 免疫细胞和胰岛β细胞
胰岛内存在一定数量的先天性免疫细胞,其中巨噬细胞占80%左右。研究发现,胰岛内先天免疫细胞可调控胰岛β细胞功能,维持机体稳态[6]。在胰管结扎小鼠模型中,M2型巨噬细胞可通过分泌转化生长因子β1,诱导胰岛β细胞中Smad7的上调,从而促进胰岛β细胞增殖[7]。相反,小鼠巨噬细胞部分清除模型中胰岛β细胞增殖明显受抑制[8],因此,巨噬细胞对于诱导胰岛β细胞增殖和更新具有重要的调控作用。Dalmas等[9]研究发现,胰岛间充质细胞分泌的IL-33作用于胰岛内驻留的第2组先天淋巴样细胞(islet-resident group 2 innate lymphoid cells,ILC2s),通过分泌IL-13和集落刺激因子2促进巨噬细胞和树突状细胞中产生视黄酸,从而诱导胰岛β细胞分泌胰岛素。目前,胰岛内的先天性免疫细胞及其信号转导促进胰岛β细胞维持更新、分泌胰岛素的详细机制尚不明确,仍需进一步深入研究。
1.3 肠道菌群和胰岛β细胞
除胰岛内驻留的先天免疫细胞对胰岛功能起调节作用外,肠道菌群对于调控胰岛功能具有重要的作用。研究发现,肠道菌来源的短链脂肪酸可激活迷走神经促进胰岛素分泌。Zhang等[10]发现肠道潘氏细胞分泌的溶菌酶可激活肠道共生菌释放肽聚糖受体1(nod like receptor1,Nod1),促进下游衔接蛋白Rip2定位至胰岛素小泡,招募Rab1a驱动胰岛素转运,促进胰岛素分泌。
1.4 常见促胰岛素分泌剂
目前临床上使用的促胰岛素分泌剂包括磺酰脲类药物和格列奈类药物。磺酰脲类药物通过与胰岛β细胞膜上的磺酰脲受体(sulphonylurea receptor,SUR)结合,阻断与其偶联的ATP敏感钾通道(ATP-sensitive potassium channel,KATP),引起钙通道开放,使胞浆内钙离子浓度升高,从而刺激胰岛素分泌。格列奈类药物同样作用于胰岛β细胞膜上的KATP,但这两类药物仅适用于胰岛功能尚存的糖尿病患者。促胰岛素分泌剂的长期使用易导致胰岛β细胞凋亡[11]。因此,寻找促进胰岛素分泌并对胰岛β细胞具有保护功能的药物作用新靶点对于糖尿病治疗意义重大。目前,临床中现有的降糖药对胰岛β细胞功能和存活作用尚不明确。本课题组通过对磺酰脲类降糖药物进行筛选,发现格列美脲显著抑制内质网应激引起的胰岛β细胞凋亡,可以有效恢复胰岛β细胞功能与体量,降低小鼠体内血糖水平。
研究发现,胰高血糖素样肽-1(glucagon like peptide 1,GLP-1)作为肠促胰岛素激素,可促进胰岛素分泌,诱导胰岛β细胞增殖,从而抑制胰岛β细胞凋亡[12]。但是,体内有活性的GLP-1易被二肽基肽酶(dipeptidyl peptidase-4,DPP4)水解,导致GLP-1失活,减弱其胰岛β细胞保护功能。基于上述机制,目前已开发GLP-1受体激动剂利拉鲁肽和DPP-4抑制剂西格列汀,均已应用于临床。
2 抑制胰岛β细胞凋亡
1型糖尿病中,胰岛β细胞特异性抗原诱导T细胞分化成熟,诱发细胞因子如IL-1β,TNF-α和NO的分泌,从而促进胰岛β细胞凋亡[13]。2型糖尿病中,糖脂毒性等可引起线粒体功能受损和ATP合成减少,减弱胰岛素分泌[14]。此外,高血糖和游离脂肪酸诱导活性氧生成,可引起内质网应激、氧化应激和炎症反应等,促进胰岛β细胞凋亡。因此,抑制胰岛β细胞凋亡可有效恢复胰岛β细胞功能和体量。
2.1 线粒体和胰岛β细胞
线粒体功能障碍可引起胰岛β细胞胰岛素分泌受阻,并可启动细胞凋亡程序。Zhang等[15]发现,高糖引起电压依赖性阴离子通道1(voltage-dependent anion-selective channel protein1,VDAC1)表达升高,VDAC1通过错误地靶向胰岛β细胞膜引起细胞中ATP损耗、细胞代谢-分泌脱偶联,降低细胞功能(图1)。研究证实,传统胰岛素增敏剂二甲双胍具有抑制VDAC1的功能。此外,在糖尿病大鼠中,蒽醌-糖苷通过抑制氧化应激反应和线粒体凋亡,改善血脂代谢,减少胰岛β细胞凋亡[16]。
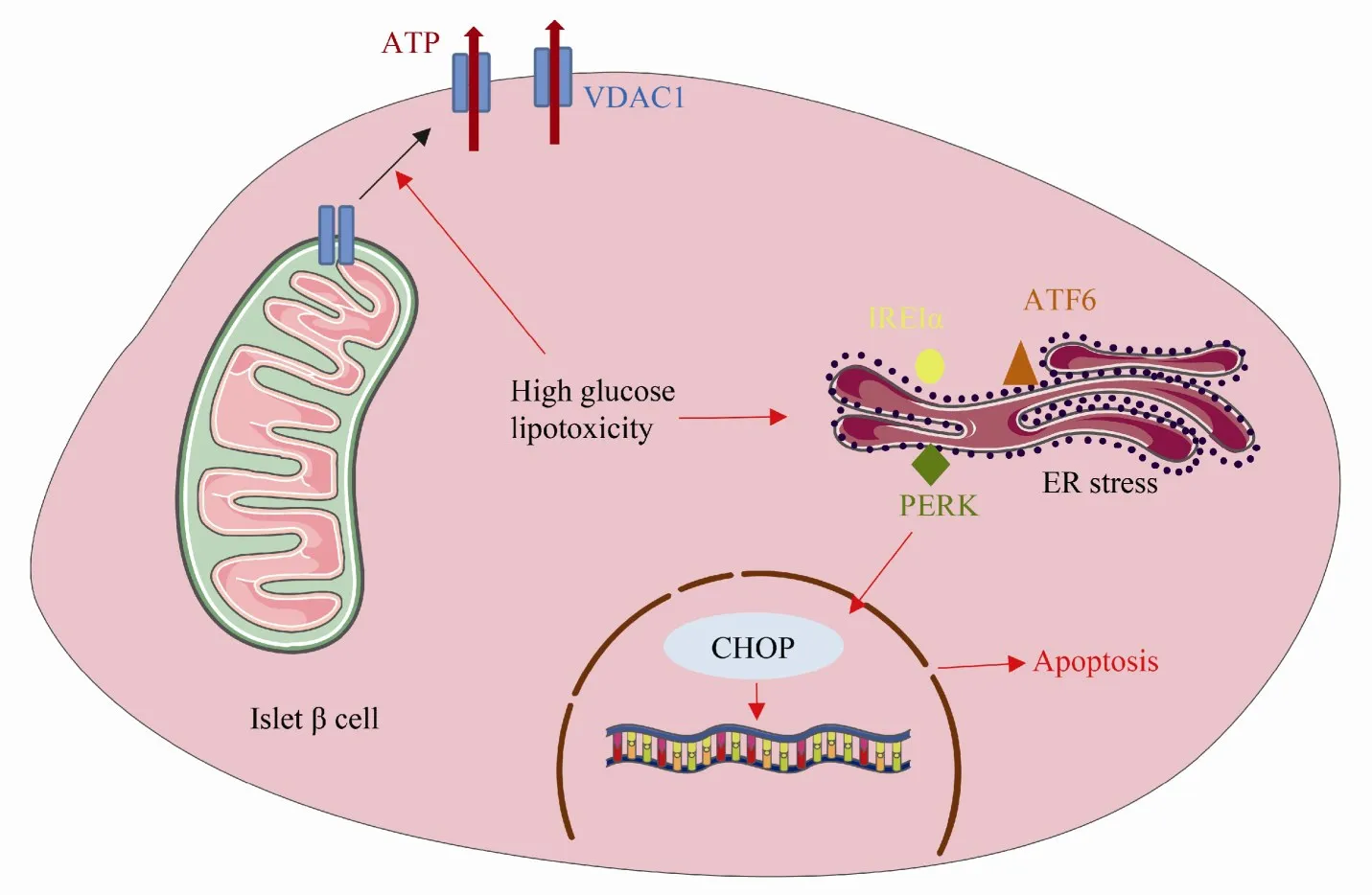
Figure 1 Mechanisms of islet β cell apoptosis in type 2 diabetes
2.2 内质网应激和胰岛β细胞
胰岛β细胞中内质网分布广泛,以维持正常的胰岛素分泌和释放[17]。高糖环境下,内质网未折叠或错误折叠蛋白增多,导致内质网功能紊乱,启动适应性信号转导,即未折叠蛋白反应(unfolded protein response,UPR)。UPR通过激活肌醇酶1α(inositol requiring enzyme 1α,IRE1α)、PKR类似的内质网激酶(PKR-like ER kinase,PERK)和激活性转录因子 6(activating transcription factor 6,ATF6)信号通路,减少蛋白合成,增强蛋白折叠能力,从而维持内质网稳态。
持续的内质网应激可引发细胞启动凋亡程序,其中C/EBP同源蛋白(C/EBP homologous protein,CHOP)作为PERK信号通路下游的转录因子发挥促凋亡作用。糖尿病小鼠模型中,CHOP敲除可显著减少胰岛β细胞凋亡,CHOP过表达则降低抗凋亡蛋白Bcl-2的表达,升高活性氧水平[18]。而且,持续性内质网应激可导致IRE1α活化,剪切X盒结合蛋白(X-box-binding protein 1,XBP1),降解胰岛素mRNA,减少胰岛素的合成和释放[19]。内质网应激过程中,敲除ATF6α的小鼠其应激敏感性增加[20]。通过高通量筛选技术,Duan等[21-22]发现2,4-二氨基喹唑啉化合物9c和3-(N-哌啶基)甲基苯甲酰胺衍生物13d(图2)均显著抑制内质网应激,恢复胰岛β细胞功能和体量,降低糖尿病小鼠血糖水平。因此,维持内质网稳态从而保护胰岛β细胞有望成为潜在的糖尿病治疗策略。
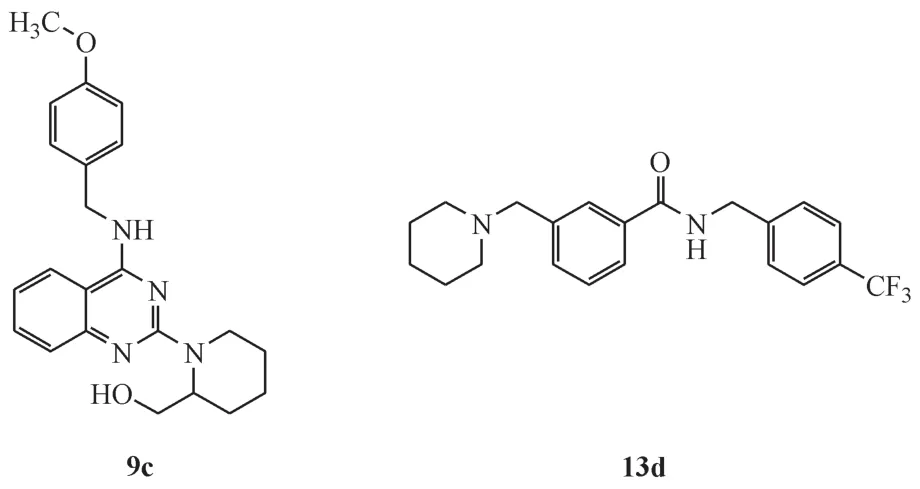
Figure 2 Chemical structures of compound 9c and compound 13d
3 促进胰岛β细胞增殖
研究报道,1型和2型糖尿病均会引起胰岛β细胞数量减少和功能减退,且随着年龄增长,成人体内胰岛β细胞的增殖能力丧失,几乎不增殖[23]。因此,促进胰岛β细胞增殖是恢复其功能和体量的重要策略之一,近年来,关于胰岛β细胞增殖调控的相关基因逐渐被发现并确证。
3.1 碳水化合物反应元件结合蛋白
碳水化合物反应元件结合蛋白(carbohydrate response element binding protein,ChREBP)作为一种调控糖脂代谢的转录因子,可与碳水化合物反应元件结合,从而驱动靶基因的表达,同时也在胰岛中表达并参与胰岛β细胞的增殖过程。研究发现,ChREBP可增加核因子红细胞系-2相关因子2(nuclear factor erythroid 2 related factor 2,Nrf2)的表达和活性,发挥抗氧化作用并诱导线粒体生物发生,促进葡萄糖诱导的胰岛β细胞增殖[24]。同时,过表达Nrf2亦可在体外诱导人胰岛β细胞增殖(图3)。
3.2 丝氨酸蛋白酶抑制剂B
研究发现肝脏分泌蛋白丝氨酸蛋白酶抑制剂B(serpin B1)促进胰岛β细胞增殖,在敲除serpin B1小鼠中胰岛β细胞增殖减慢,而在斑马鱼中过表达serpin B1引起胰岛β细胞增殖。在肝脏特异性敲除胰岛素受体引起胰岛素抵抗的小鼠模型中,敲除肝脏叉头框转录因子O1(forkhead box protein O1,FoxO1)可降低serpin B1的表达,从而抑制胰岛β细胞增殖[25],但serpin B1的下游分子机制尚不明确(图3)。
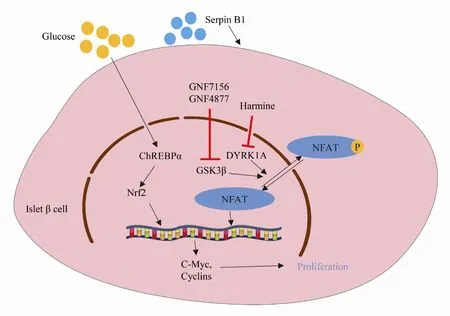
Figure 3 Pathways involved in islet β cell proliferation
3.3 双底物特异性酪氨酸磷酸化调节激酶A
双底物特异性酪氨酸磷酸化调节激酶A(dualspecificity tyrosine-phosphorylation regulated kinase 1A,DYRK1A)是一种进化上高度保守的蛋白激酶。DYRK1A可通过磷酸化细胞周期相关蛋白如P21和Cyclin D1调控细胞的增殖和分化过程。Wang等[26]筛选发现,在不同糖尿病动物模型中,骆驼蓬碱类似物均可抑制calcineurin-NFATDYRK1A信号通路促进胰岛β细胞增殖,从而控制血糖水平。Shen等[27]亦发现氨基吡嗪类化合物GNF7156可抑制DYRK1A和糖原合酶激酶3β(glycogen synthase kinase-3,GSK3β)的活性,促进人胰岛β细胞增殖,改善糖尿病小鼠血糖水平。以上结果提示抑制DYRK1A的活性具有促进胰岛β细胞增殖的作用(图 3)。但Rachdi等[28]发现,在敲除DYRK1A小鼠中,葡萄糖耐量降低,胰岛β细胞增殖以及胰岛β细胞的体量均减少。提示在不同种属中,DYRK1A在胰岛β细胞中发挥的生物学作用可能有所不同,以DYRK1A为靶点的药物研究仍需进一步探讨。
4 诱导其他种类细胞分化成为胰岛β细胞
诱导其他种类细胞分化为具有细胞功能且表达成熟胰岛β细胞标志基因NEUROD1,NKX6.1,PAX6,PDX1,MafA的胰岛β样细胞是增加胰岛β细胞体量和保护胰岛β细胞功能的另一重要方法[29]。目前研究热点主要包括诱导胰岛α细胞或胰腺导管细胞、胰腺前体细胞和腺泡细胞以及干细胞分化为胰岛β样细胞等方面,其中对干细胞进行诱导分化的研究最为深入。
在体外,Pagliuca等[30]利用诱导性多能性诱导干细胞(induced pluripotent stem cells,iPSC)诱导产生具有葡萄糖反应性的成熟β样细胞,该诱导细胞可表达成熟胰岛β细胞的标志物PDX1,调节胰岛素分泌,改善小鼠的血糖水平。Veres等[31]发现用带有标记CD49a抗体的磁珠可准确富集胰岛β样细胞,提高细胞分化效率。
除多能干细胞外,其他来源的干细胞亦可诱导分化为胰岛β样细胞。2016年,实验发现糖尿病患者的表皮细胞可转分化成iPSC,再进一步分化为具有葡萄糖应答反应的胰岛β样细胞[32]。此外,Saxena等[33]采用基因编程技术,选取一位50岁受试者提取其脂肪组织来源的间充质干细胞,成功分化成为功能性胰岛β细胞。Sancho等[34]发现抑制SCF型泛素连接酶组成成分FBW7的活性,可诱导胰腺导管细胞分化成为胰岛β样细胞。Lin等[35]发现在胚胎干细胞(embryonic stem cells,ESC)中转染PAX4可显著提高ESC分化为胰岛β样细胞的效能。在斑马鱼中进行全生物筛选发现,双硫仑或霉酚酸可分别通过抑制视黄酸的合成或降低细胞的GTP水平诱导前体细胞分化为胰岛β样细胞[36]。目前,定向诱导多种来源的细胞分化为具有分泌功能性胰岛β样细胞依然存在相关技术及理论瓶颈,但以胰岛β细胞再生为策略的糖尿病治疗手段依然是基础和临床研究关注重点。
5 结 语
随着对糖尿病研究的不断深入,胰岛β细胞的功能和体量在糖尿病的发生发展中的作用也逐渐受到关注。目前已发现许多新型天然产物可改善血糖水平[37-38]。此外,基于胰岛β细胞保护对临床使用的降糖药物进行研究也有助于药物的合理应用。近年来,随着免疫系统和肠道微生物调控胰岛功能作用的深入研究,关于胰岛β细胞增殖的机制逐渐被阐明。高糖高脂及炎症信号等病理刺激下导致胰岛β细胞凋亡的相关研究也不断推进。促进胰岛β细胞增殖及诱导其他细胞分化为胰岛β细胞是提高胰岛β细胞体量的有效方法,但是通过药理学诱导胰岛β细胞增殖的方式仍需要对相关药物的致癌性进行评估,同时关注血液循环中胰岛素及血糖水平,以保证药物的安全性。
在糖尿病治疗中,恢复胰岛β细胞体量和功能的策略日益受到关注。目前大量基础研究、药物筛选以及相关的干细胞疗法研究不断深入,同时取得了一定的成果,展现出良好的前景。但是,此策略是否能运用于临床,服务于糖尿病患者还需要多学科配合系统地展开研究。
猜你喜欢
清华金融评论(2022年4期)2022-04-13 21:33:11
解放军医学杂志(2021年12期)2022-01-18 03:53:24
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:46
现代临床医学(2021年1期)2021-01-26 00:55:52
生命科学研究(2018年1期)2018-05-29 01:13:01
安徽医科大学学报(2016年12期)2017-01-15 14:21:55
中国当代医药(2015年33期)2015-03-01 02:09:08
肝胆胰外科杂志(2015年1期)2015-02-27 11:11:34
中国药理学通报(2014年2期)2014-05-09 08:22:15
中国医学科学院学报(2014年6期)2014-03-11 20:26:19
