
Tumor microenvironment in primary liver tumors: A challenging role of natural killer cells
2020-10-29 02:03MichelaAnnaPolidoroJoannaMikulakValentinaCazzettaAnaLleoDomenicoMavilioGuidoToriliMatteoDonadon
World Journal of Gastroenterology 2020年33期
Michela Anna Polidoro, Joanna Mikulak, Valentina Cazzetta, Ana Lleo, Domenico Mavilio, Guido Torili,Matteo Donadon
Abstract In the last years, several studies have been focused on elucidate the role of tumor microenvironment (TME) in cancer development and progression. Within TME, cells from adaptive and innate immune system are one of the main abundant components. The dynamic interactions between immune and cancer cells lead to the activation of complex molecular mechanisms that sustain tumor growth. This important cross-talk has been elucidate for several kind of tumors and occurs also in patients with liver cancer, such as hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA). Liver is well-known to be an important immunological organ with unique microenvironment. Here, in normal conditions, the rich immune-infiltrating cells cooperate with non-parenchymal cells, such as liver sinusoidal endothelial cells and Kupffer cells, favoring self-tolerance against gut antigens. The presence of underling liver immunosuppressive microenvironment highlights the importance to dissect the interaction between HCC and iCCA cells with immune infiltrating cells, in order to understand how this cross-talk promotes tumor growth. Deeper attention is, in fact, focused on immune-based therapy for these tumors, as promising approach to counteract the intrinsic anti-tumor activity of this microenvironment. In this review, we will examine the key pathways underlying TME cell-cell communications, with deeper focus on the role of natural killer cells in primary liver tumors, such as HCC and iCCA, as new opportunities for immune-based therapeutic strategies.
Key words: Primary liver cancer; Natural killer cells; Tumor microenvironment; Hepatocellular carcinoma; Intrahepatic cholangiocarcinoma; Immune cells
INTRODUCTION
Tumor microenvironment (TME) has emerged as a pivotal factor in driving tumor development and progression[1]. Cells from both adaptive and innate immune system are the main components of TME, which establishes dynamic interactions with cancer cells. The resultant cross-talk leads to activation of complex molecular mechanisms that finally foster tumor growth by inhibition of anti-tumor activity of immune cells[2]. This phenomenon occurs also in patients with liver tumors, such as hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA). Being, the liver an important organ, in which the rich immune-infiltrating cells with non-parenchymal cells, such as liver sinusoidal endothelial cells and Kupffer cells (KCs) cooperate to maintain the immunosuppressive microenvironment favoring self-tolerance against gut antigens, it is of paramount importance to know how HCC and iCCA cells interact with immune infiltrating cells to promote the pro-inflammatory and immunosuppressive environment that finally foster tumor growth. In this narrative review, we explore the key pathways involved in TME cell-cell communications, with particular focus on the emerging role of natural killer cells (NK) as new opportunities for immune-based therapeutic strategies. For doing that, a comprehensive literature search was conducted using PubMed to identify relevant articles published between 2000 and 2020. The search was limited to articles in English and it was further broadened by extensive cross-checking of all the references in the articles retrieved to identify eventual additional non-indexed literature.
LIVER IMMUNOSURVEILLANCE
In physiological conditions, the liver has a unique microenvironment in which a delicate balance between cells of the innate and adaptive immune systems is required to maintain a strong immunosuppressive microenvironment[3,4].
Daily, about 80% of liver blood flow derives from the gastrointestinal tract through the portal vein carrying high concentrations of pathogen-derived molecules. Due to this high load of bacterial antigen, liver immunosurveillance plays a crucial role in maintaining self-tolerance, thus avoiding a severe immune self-response[5,6].
There are many cell populations involved in the hepatic tolerogenic process. The first line of defense is represented by liver sinusoidal endothelial cells (LSECs), the most abundant non-parenchymal liver cells with scavenger and immunologic functions. These unique cells have a high expression of several scavenger receptors, such as mannose receptors, major histocompatibility complex (MHC) class I (MHC-I) and MHCII[7]. These surface receptors allow for internalization of the antigens of pathogens, presenting them directly to T lymphocytes. Their role as antigen presenting cells (APCs) together with the increasing expression of co-inhibitory molecules, such as programmed death-ligand 1 (PD-L1) on LSECs, after recognition of antigens drives CD8+ T cell tolerance. Moreover, LSECs have been shown to influence APC functions of dendritic cells (DCs), leading to a reduction in their ability to activate T cells[8,9].
KCs are tissue resident macrophages located throughout the liver sinusoids. KCs have phagocytic and cytokine secretion activities, thus eliminating circulating molecules and releasing IL-10 and transforming growth factor beta (TGFβ), which leads to suppression of T cell activity[10,11].
Furthermore, the liver is rich with innate immune cells, including NK and NK T cells, and cells from the adaptive immune system, such as T and B lymphocytes. Under a steady-state condition, the balance of these cell functions is crucial for preventing the acute immune response within the liver against common gut pathogens.
Emerging role of TME
In the last two decades, tumorigenesis has been recognized as a complex and dynamic process orchestrated by multiple different cell types, with each one of them playing a key role in tumoral development and progression[12].
Despite cancer cells, which have developed as a consequence of genetic mutations, holding the main role in driving carcinogenesis, an increasing interest has been aimed to TME, pointed out as a contributor to progression and metastatization of several tumors[13,14].
Cancer-associated fibroblasts (CAFs), blood and lymphatic vascular network, extracellular matrix and immune cells from both innate and adaptive immunity are principal components of TME (Figure 1)[15]. In normal conditions, the main role of immune surveillance is protection against pathogens, maintenance of tissue homeostasis and eradication of incipient cancer cells[16,17]. In contrast, in sites of chronic inflammation, as in neoplasia, immune inflammatory cells could persist and display an aberrant effect on cancer cells.
Immune cells could be present at any sites of tumor, from the center to the invasive margin, and their location or density have been largely demonstrated having a chief regulatory effect in promoting tumor progression[18,19]. These immune cells sculpt the TME through the secretion of several molecules, as growth factors, cytokines and chemokines, which in turn sustain and augment this inflammatory state, stimulating cancer cell proliferation, tumor angiogenesis and spreading[20-22].
Furthermore, the TME has been described to be a prognostic factor for several tumors and involved in the response of tumors to conventional therapies[22,23]. This recent comprehension of a tumor as a multi-cellular complex has allowed us to understand the mechanism underlying the crosstalk between cancer and tumorinfiltrating immune cells to design immune-based therapy[24].
Most of the research on therapies targeting the TME have been extensively focused on enhancing T cell cytotoxic activities. Immune checkpoint inhibitors, as antagonists of programmed cell death protein 1 (PD-1), PDL-1 and cytotoxic T lymphocyte antigen 4 (CTLA4), have been promising and efficacious in ameliorating patient prognosis with different solid tumors and hematological malignancies[25-27].
On the other hand, from innate immunity, NK cells are gaining more attention. These cells have been recognized to have a role in immune surveillance by excreting cytotoxic substances that eliminate malignant cells[28]. Studies highlighted the positive correlation between NK cell infiltration in tumors and a better prognosis[29,30]. Despite that, in tumors, they exhibit low to no cytotoxic activity, due to the immunosuppressive environment of the TME[31,32].
Efforts in identifying of mechanism aimed to restore the anti-tumoral effect of NK cells could represent the basis for developing new immune-based therapeutic strategies, leading to more effective treatments in combination with conventional therapies[33-35].
NK cells: Key features in healthy liver
Since their discovery, NK cells have been valued for their rapid recognition and clearance of tumor cells without previous stimulation and antigenic specificity[31,36].
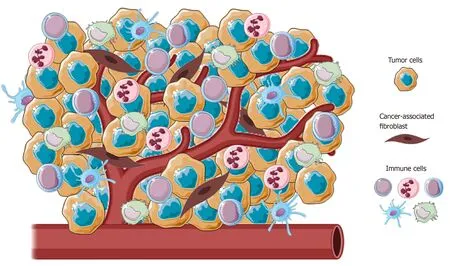
Figure 1 Representative image of the main cellular component of tumor microenvironment. Tumors present a wide array of cells from both innate and adaptative immunity (i.e., macrophages, natural killer cells, neutrophils and lymphocytes) able to foster cancer growth and malignancy.
NK cells are able to kill tumor cells through several mechanisms, including exocytosis of cytotoxic perforin and granzymes; tumor necrosis factor (TNF) family death receptors, such as fas ligand (FasL) or TNF-related apoptosis-inducing ligand (TRAIL); antibody-dependent cellular cytotoxicity; and pro-inflammatory cytokine release, such as interferon (IFN)-γ[37]. In healthy adults, NK cells represent about 10%-15% of circulating lymphocytes that are subdivided into two distinct subsets defined on the basis of the cellular membrane expression of CD56 and CD16, namely, CD56bright(CD56brightCD16neg) and CD56dim(CD56dimCD16pos)[37,38]. The two CD56brightand CD56dimNK cell subsets are distinct in their tissue distribution and their roles in immunity. Mainly represented in the blood (up to 90%), the CD56dimNK subset primarily acts through their high cytotoxic activity, although growing evidence shows their capability to also produce IFN-γ[39]. On the other hand, the CD56brightNK subset is less cytotoxic but exerts important immune-regulatory functions through secretion of chemokines and pro-inflammatory cytokines (i.e., IFN-γ and TNF-a) in response to different stimuli (i.e., IL-1β, IL-2, IL-12, IL-15 and/or IL-18) delivered by surrounding cells at tissue sites (i.e., macrophages, DCs and T lymphocytes)[36,40,41]. In fact, the CD56brightsubset represents only 5-10 % of circulating NK cells and mainly resides in peripheral tissues such as liver, and gastrointestinal and female reproductive tracts. Although it is still being debated, CD56brightNK cells are largely accepted as precursors of more differentiated CD56dimNK cells[42].
The activation of NK cells is controlled by an array of inhibitory and activating NK cell receptors (iNKRs and aNKRs, respectively) differently expressed at their cell surface[43]. In resting conditions, NK cell cytotoxic activity is repressed due to the inhibitory receptors, including inhibitory killer Ig-like receptors (KIRs) and the C-type lectin receptor NKG2A, recognizing alleles of MHCI. Based on the “missing selfhypothesis”[44], the absence of MHC-I on target/tumor cells gives “license” to NK cell killing through aNKRs, such as natural cytotoxicity receptors (NCRs; NKp30, NKp46, and NKp44), the C-type lectin receptors NKG2D and NKG2C, DNAX accessory molecule-1 (DNAM-1) and activating KIRs (aKIRs) that bind their putative ligands on stressed, viral infected or tumor cells[45].
Human liver resident NK (lr-NK) cells were described for the first time in the late 1970s and defined as highly cytotoxic NK cells resident in the hepatic sinusoids[46,47]. Differently to peripheral blood, CD56dimand CD56brightNK cells are present at similar frequencies in human liver[48,49]. Recently, the specific phenotype of human CD56brightlr-NK cells has been described with the constitutive expression of the chemokine receptors CXCR6 and CCR5, along with the tissue-residency marker CD69[48,50,51]. The mechanisms involved in the recruitment of NK cells at the liver site are still unclear, although the interaction of NK cells with sinusoidal endothelial cells certainly plays a key role. Additionally, high expression of CXCR3, CXCR6 and CCR5 on lr-NK cells may play an important role in their liver retention, since their cognate ligands (i.e., CCL3, CCL5 and CXCL16) are highly produced by cholangiocytes, sinusoidal endothelial cells, hepatocytes and KCs[48]. Lr-NK cells are characterized by strong cytotoxic activity, high constitutive expression of TRAIL and FasL and secretion of IFN-γ, TNF-a, granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-2[52-54]. On the other hand, the human liver has developed a high degree of immune tolerance[55]. In order to maintain a tolerogenic liver environment, KCs produce high doses of IL-10 that are critical in the control of NK cell-mediated alloreactivity[56,57]. Moreover, the interplay between lr-NK cells and DCs induces the expansion of tolerogenic T cells (T regs) via the engagement of the inhibitory NKG2A receptor[58]. Lr-NK cells are also important in liver regeneration after tissue damage[59,60]. The interaction of NK cells with KCs, fibroblasts and stem cells induces the secretion of several soluble factors able to induce the proliferation of hepatic cells[59,61], although overstimulation of lr-NK cells can inhibit, rather than promote, liver regeneration[62].
Interestingly, in the liver, lr-NK cells have been identified to be endowed with a unique so-called “memory-like” NK (ml-NK) phenotype[63]. Human ml-NK cells have been described in cytomegalovirus (CMV) infection, resulting in the specific NKG2C+ phenotype able to produce a higher amount of IFN-γ upon being re-challenged with the same virus[64,65]. However, the existence of these ml-NK cells in other human virus infections, such as hepatitis, have not been reported yet. Thus, the impact of the liver in generation of highly heterogenous lr-NK cell subsets reflect either their cytotoxic or tolerogenic profiles.
HCC
Introduction
HCC is the first liver tumor and the third leading cause of cancer-related death worldwide[66,67]. The majority of HCC occurs in patients with underlying liver disease. Patients with chronic liver disease have sustained hepatic inflammation, fibrosis and aberrant hepatocyte regeneration[68]. Specifically, there are some clearly defined agents related to cancer development, including HBV, HCV, metabolic causes (non-alcoholic fatty liver disease and nonalcoholic steatohepatitis) and external factors (e.g., aflatoxin)[69-71]. Timing of HCC diagnosis is crucial for patient life expectancy[72]. At the early stage, surgical resection, transplantation or local ablation have been demonstrated to improve clinical outcomes in patients with HCC, despite a high recurrence rate of about 70% at 5 years[73-76].
Unfortunately, more than 50% of newly diagnosed patients already have advanced or unresectable disease. For these patients, prognosis and treatment are very challenging, in particular when underlying liver dysfunction could limit most of the available therapeutic options.
Outcomes with traditional chemotherapies have been investigated in several clinical trials with no statistically significative improvement in OS[77-79]. Lack of response is notoriously described for HCC and may be due to the chemo-refractoriness of hepatocytes, which are able to express a variety of multi-drug resistance genes and p53 mutations[80,81]. Moreover, cytotoxic therapies may be limited in the setting of advanced stage disease if underlying liver cirrhosis is present[82]. This highlights that the development of new therapies is fundamental for the management of patients with HCC. In the last few year, increasing attention has been focused on target therapy to develop a more effective treatment for patients with HCC[83].
In 2007, sorafenib was approved as a first-line treatment for patients with advanced or metastatic HCC, showing an increase in the OS of patients compared to standard treatment[84-87]. Sorafenib, is a multi-kinase inhibitor with anti-proliferative and antiangiogenic effects, increases HCC cell apoptosis by blocking several molecular targets, including Raf/MEK/ERK pathway, vascular endothelial growth factor receptor (VEGFR)-2, VEGFR-3, platelet-derived growth factor receptor beta (PDGFR-β) and many other tyrosine kinases[88,89]. Despite such efforts, the median life expectancy of patients with HCC treated with sorafenib is about 1 year[90].
In these patients, an adverse scenario, immunotherapy, is gaining a relevant role as a potential tool for new immune target strategies focused on counteracting the immunosuppressive HCC microenvironment.
TME in HCC
HCC has been recognized to be an immunogenic cancer, arising from chronic liver inflammation, as a result of viral infection or toxin[91,92]. Several studies have shown that this tumoral milieu, which is also enriched in several pro-inflammatory chemokines released by tumoral and non-tumoral cells, enhances the immunosuppressive physiological microenvironment contributing to HCC progression[93,94].
As a result of the persistent inflammatory state, HCC has a rich immune infiltrate, in which tumor-infiltrating lymphocytes (TILs) represent one of the most abundant populations within the TME[95]. Tumor-associated Treg cells have been shown to have a detrimental impact, favoring tumor immune evasion by markedly reducing the activity of effector cells through secretion of both IL-10 and TGFβ and cell-cell interaction[96].
Moreover, the inhibitory PD-L1 molecule was found to be highly expressed in patients with HCC from KCs, tumor cells and LSECs correlating with PD-1 upregulation on CD8+ cytotoxic cells. These mechanisms lead to the dysfunctional activity of CD8+ cells resulting in an exhausted phenotype (Figure 2).
Of note, the balance between Treg and cytotoxic T cells infiltration has been found to correlate with the prognosis. A higher presence of CD4+ cells in tumoral areas correlate with increased recurrence risk and worse OS[97-99]. In this regard, the overexpression of immune checkpoints in cancer and TME cells and their related pivotal role in HCC may represent a promising therapeutic strategy for counteracting anti-tumor immunity.
To date, several clinical trials are ongoing to investigate the efficacy of blockade of three immune checkpoints (anti-CTLA-4 and anti-PD-1/PD-L1) as a single agent or in combination with standard therapies for the treatment of advanced HCC[100]. Besides the promising results observed in these clinical trials, increasing attention has been focused on NK cells to develop a new immune strategy. Their role, localization and future perspective in HCC are discussed in detail below.
Role of NK cells in HCC
Hepatic NK cells are thought to play an important role in the immunological protection against different liver cancers, including HCC[52,53,101,102]. However, several numerical and phenotypic changes have been described in NK cells during the development of HCC. Patients with HCC at various stages of disease show both reduced frequencies and absolute number of peripheral blood NK cells. In particular, the specific CD56dimNK cell subsets displayed a dramatic reduction in patients with HCC[103,104]. Studies focused on intrahepatic NK cells in patients with HCC also observed the reduction in the frequency of tumor-infiltrating NK cells compared to tumor-adjacent lr-NK cells, which was mainly related to the reduced number of the CD56dimNK cell subset[103,105]. However, a higher number of total CD56+ (CD56dimand CD56bright) tumor- infiltrating NK cells predict a better outcome with regard to OS in patients with HCC[106,107].
In addition, some studies have reported the increased frequency of specific NK cell subsets associated with slower HCC progression, as was observed for CD11b-/CD27- NK cells[108]. Regarding the specific molecular mechanism that boosts anti-tumor NK cell activity, the engagement of NKG2D activating receptor has been shown to enhance NK cell cytotoxicity against HCC[109]. However, this potent anti-tumor NK cell effector-function against HCC seems to be more effective in the early stages of HCC and decreases as soon as the tumor progresses. In fact, the reduced frequency of both circulating and intrahepatic NK cells was particularly noticeable in patients with advanced stages of HCC[110]. Moreover, both CD56dimand CD56brightNK cells in patients with end-stage HCC exhibited anergic effector functions in the peripheral blood and at the tumor site[103]. Specifically, reduced NK cell cytotoxic activity (i.e., lower production of granzymes and cytotoxic perforin) and lower secretion of cytokines (i.e., TNF-a and IFN-γ) associated with the progression and invasion of HCC were reported[111,112]. Various mechanisms are involved in the functional impairment of NK cells in advanced HCC[113]. For instance, down-modulation of NKG2D results in defective NK cell activation and recognition of tumor cells[109,114,115].
On the other hand, excessive stimulation of numerous inhibitory receptors expressed on NK cells negatively control their anti-tumor response. In particular, expression of the specific inhibitory NKp30 splice-variant along with higher levels of its soluble ligand (NKp30L) B7-H6 were found in patients with the late stages of HCC[115]. The aberrant engagement of the NKp30 pathway and CD48/2B4 interaction with tumor-infiltrating macrophages also induce rapid NK cell exhaustion[110,111,116]. Moreover, the specific immunogenetic profile of KIR/HLA affects the prognosis of patients with HCC[117]. Likely, high expression of HLA-E molecule in HCC triggers the inhibitory NKG2A receptor[118]. Albeit KIRs and NKG2A, several other immune checkpoints, including PD-1, Tim-3 and CD96, can inhibit the activity of NK cells in HCC[119]. Poor clinical outcomes for patients with HCC correlates with expression of PD-1 and CD96 on tumor-infiltrating NK cells[120,121](Figure 3).
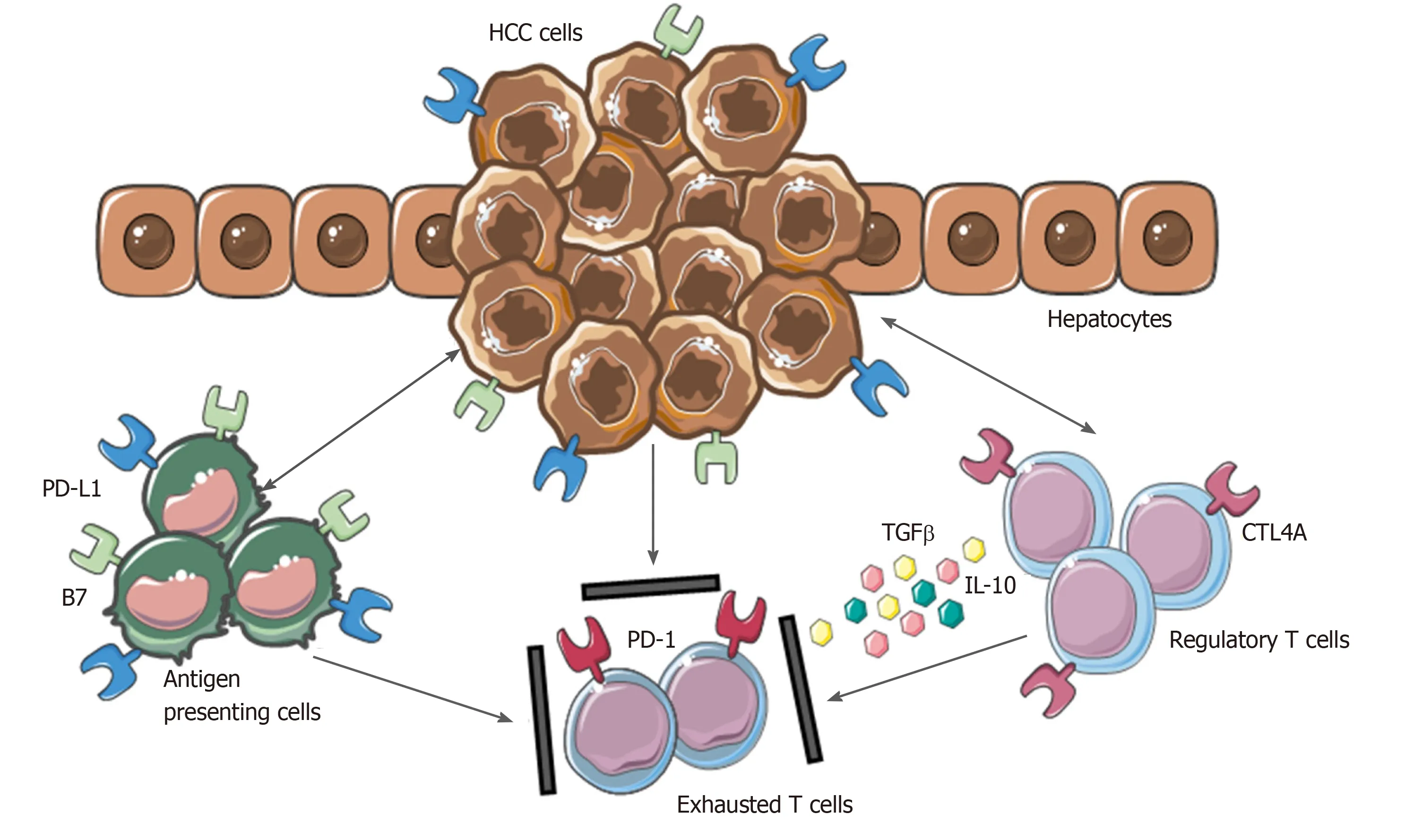
Figure 2 Mechanisms involved in hepatocellular carcinoma immune evasion. In physiological conditions, liver has the ability to induce immunotolerance against antigen from gastrointestinal tract. These mechanisms have a detrimental role during hepatocellular carcinoma development and progression. Upregulation of inhibitory programmed death-ligand 1 molecule from tumor cells, Kupffer cells, liver sinusoidal endothelial cells and antigen presenting cells, together with the release of interleukin-10 and transforming growth factor beta, lead to an exhausted phenotype of CD8+ cells and prevent tumor cells from immune damage. HCC: Hepatocellular carcinoma; PD-L1: Programmed death-ligand 1; CTL4A: Cytotoxic T lymphocyte antigen 4; PD-1: Programmed cell death protein 1; TGFβ: Transforming growth factor beta; IL-10: Interleukin-10.
Additionally, the increased expression of PD-1 and Tim-3 on NK cells was found to significantly increase during chronic HBV and HCV infections[97,122]. Another mechanism contributing to NK cell impairment in HCC relies on the expansion of CD4+/CD25+ T regs and increased secretion of the immunosuppressive cytokines, including IL-10 and TGFβ[110,111,123]. The variations in the cytokine milieu able to inhibit cytotoxic activity and secretion of cytokines by NK cells in HCC also include soluble immunomodulators, such as TGFβ, prostaglandin E2 (PGE2) and indoleamine 2,3-dioxygenase (IDO)[124,125].
Numerus strategies employed by HCC to evade NK cell immunosurveillance in later stages of the disease are used for HCC treatment[112,113]. Molecular target drugs such as sorafenib, a multikinase inhibitor, and bortezomib, a proteasome inhibitor, trigger hepatic NK cell antitumor responses, resulting in higher cytotoxicity and IFN-γ production[126]. In addition, histone deacetylase inhibitors (HDACis) promote MICA or MICB expression on hepatoma cells, thus increasing the susceptibility of hepatoma cells to NK cell-mediated lysis[127,128]. Recently, therapeutic strategies have been focused on targeting NK cell checkpoints, such as NKG2A, KIRs, PD-1 and CTLA4 to boost activation and reverse dysfunction in NK cells[129]. In addition, several clinical studies have demonstrated the efficacy of allogenic NK cells in adoptive immunotherapy of HCC treatment[130].
ICCA
Introduction
CCA accounts for about 15% of all primary liver malignancies and is second after HCC in number of cases[131,132]. Indeed, CCA is a group of heterogeneous tumors arising at different levels of the biliary tree.
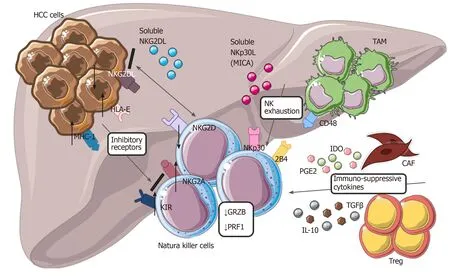
Figure 3 Modulation of natural killer cells cytotoxic activity in hepatocellular carcinoma. Tumor cells with tumor-associated macrophages and other cells within tumor microenvironment are involved in dysfunctional activity of natural killer cells (NK), reducing their ability to recognize and eliminate malignant cells. Down regulation of NKG2D, up-regulation of different inhibitory receptors, secretion of cytokines from cancer-associated fibroblasts and Treg, interaction of CD48/2B4 are the main mechanisms involved in NK exhaustion. HCC: Hepatocellular carcinoma; NK: Natural killer cells; CAF: Cancer-associated fibroblasts; TAM: Tumor-associated macrophages; IL-10: Interleukin-10; MHC-1: major histocompatibility complex class I; KIR: Killer Ig-like receptors.
The most recent classification describes three different CCA subtypes looking at different anatomical regions: iCCA, peri-hilar CCA (pCCA) and distal CCA (dCCA)[133,134]. Several studies in the last two decades revealed that the incidence of these subtypes is vary. iCCA incidence has been increasing in contrast with the decrease of pCCA and dCCA incidence[135-137].
Different risk factors and survival rates seem to be related to each of them. Despite the etiologies remaining unclear in most cases of CCA, some risk factors are well established. For example, liver flukes (Opistorchis viverriniandClonorchis sinensis) have been clearly associated with iCCA in East Asia. On the other hand, especially in Europe, primary sclerosing cholangitis (PSC) is a demonstrated risk factor for CCA, specifically correlated to pCCA variant. Viral hepatitis (HBV and HCV) has been identified as definitive risk factors more associated with iCCA than pCCA. Emerging role have been given to metabolic syndrome, alcohol and smoking[138-140].
Diagnosis is usually tardive due to its vague symptoms. Patients with iCCA are generally asymptomatic (20%-25% incidental finding), appearing tardively cachectic, with abdominal pain and fatigue. In contrast, pCCA most frequently manifests as painless jaundice. No proper biomarkers are available and diagnosis is a combination of clinical, radiological and unspecific histologic-biochemical markers. It has long been argued that a staging system for CCA needs to be found[133,141,142].
iCCA treatment is very dismal due to the well-known lack of response to the conventional chemotherapy[143,144]. Nowadays, surgical resection together with liver transplantation, for highly selected patients, are the only potentially curative treatments for iCCA, with median disease-free survival (DFS) duration of 12–36 mo. For advanced stage and unresectable iCCA, transarterial chemoembolization (TACE) is a treatment option considered to prolong OS in patients[145,146].
For patients with iCCA, due to the lack of effective curative strategies, understanding the molecular mechanism together with the TME interactions involved in tumoral progression and chemoresistance could open the possibility of developing new potential target therapies.
TME in CCA
CCA is characterized by a dense desmoplastic TME composed of high stromal cell infiltration together with immune cells from the adaptive and innate immune systems[147]. In the last years, several studies have been focused on understanding the mechanism underlying the interplay between stromal cells and neoplastic cells in CCA.
In the liver, CCA CAFs have been shown to have multiple sources of origin, including from hepatic stellate cells, circulating bone marrow-derived precursor cells and portal fibroblasts[148,149].
In the tumoral milieu, CCA cells have been reported to be the main cellular component secreting PDGF-DD, which acts by binding to its receptor PDGFR-β expressed on CAFs, leading to their recruitment in the tumor site, together with other factors, such as TGFβ[150,151].
Once recruited and activated within the tumor, CAFs are able to enhance CCA growth and progression through the secretion of tumor matrix, providing the scaffold and the release of various soluble factors[152-154].
PDGF-BB is an important paracrine survival signal released by CAFs that influence tumor malignant phenotype. By binding to its cognate receptor, PDGFR-β on CCA cells, it leads to an intracellular signaling cascade able to protect tumor cells from TRAIL-induced apoptosis by activating Hedgehog signaling and sustaining TGFβ secretion[155,156].
Among cytokines, chemokines and growth factors released by CAFs, stromal cellderived factor-1 (SDF)-1 and heparin-binding EGF-like growth factor (HB-EGF) have key roles in promoting proliferation, migration and invasion of CCA cells expressing their related receptors, CXCL4 and EGFR. In the meantime, the secretion of HB-EGF by CAFs is sustained by a TGFβ feedback release from CCA cells, upon EGFR activation[157-159].
This highlights the presence of multiple paracrine signals exchanged between CCA cells and CAFs. The latter promotes tumor cell proliferation and invasion and in turn actively recruited and activated by CCA in a self-perpetuating loop (Figure 4).
Due to key role of stromal cells in CCA TME, in the last few years, several studies have been focused on targeting CAFs and their released factors[160-162]. The most recent study investigated the role of an FDA-approved anti-fibrotic drug called nintedanib. This tyrosine kinase inhibitor is able to inhibit fibroblast growth factor receptor (FGFR) and PDGFR, showing promising results in reduction of CCA growth and aggressiveness bothin vitroandin vivo[163]. The emerging important role of CAFs could be an attractive target to ameliorate the treatment of patients with CCA.
Together with CAFs, cells from the innate and adaptive immune systems have been found to infiltrate the TME and significantly sustain CCA malignant transformation. Among them, tumor-associated macrophages (TAMs) represent the most relevant primary immune cells.
Macrophages could be resident (KCs) or recruited from circulating monocytes via soluble factors, such as monocyte chemoattractant protein 1 (MCP-1/CCL2), released in the tumor milieu[151,164]. Once in the liver, they differentiate into tissue macrophages, acquiring specific subset and activation status according to the exposition of multiple signals in the TME[165].
Normally, macrophages could be polarized in two possible phenotypes: M1 (antitumorigenesis), which is activated by INF-g, and M2 (pro-tumorigenesis) in response to anti-inflammatory cytokines, such as IL-10, TGFβ and IL-4[166,167].
In CCA TME, studies revealed that either CAF or CCA cells are involved in TAM recruitment and inducement of a M2 phenotype via the STAT3 pathway. TAMs polarized as pro-tumorigenic, the most representative TAM in TME, have been shown to promote tumor progression and modulation of the surrounding microenvironment through the subsequent secretion of pro-inflammatory and tumor-promoting mediators (IL-6, IL-1, TGFβ, VEGF and PDGF)[147,168,169]. Indeed, increasing evidence has shown that, in iCCA, the higher number of M2 TAMs correlated with a poor prognosis[170-172].
In the last year, more attention has been paid to neutrophils, the most abundant of the white blood cells, which represent one of first lines of defense against invading pathogens. Different studies have revealed that neutrophils are recruited within tumors driven by CXCL5, a chemotactic cytokine. Tumor-associated neutrophils (TANs) and elevated pre-operative neutrophil to lymphocyte ratio (NLR) are associated with a poor prognosis in patients with iCCA[173-176].
Further studies are needed to deeper elucidate the role of TAMs and TANs in iCCA, in terms of possible use as predictive markers for patients undergoing surgical resection. As described for HCC, iCCA arises in a chronically inflamed liver as a consequence of bile duct injury and the related TME contributes to the development of an anti-tumor immune milieu.
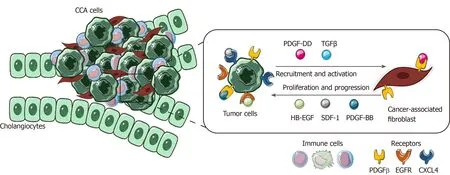
Figure 4 Role of cancer-associated fibroblasts in cholangiocarcinoma. In the figure is shown the paracrine loop between tumor cells and cancerassociated fibroblasts, the main cellular component of cholangiocarcinoma tumor microenvironment (TME). Several survival and recruitment signals are exchanged within TME, leading to tumor growth and progression. CCA: Cholangiocarcinoma; TGFβ: Transforming growth factor beta; PDGF-β: Platelet-derived growth factor receptor beta; EGFR: Endothelial growth factor receptor.
CCA cells with CAFs and TAMs are able to secrete various factors, such as CCL2, recruiting and stimulating T regs. These release back IL-10 and TGFβ that inhibit cytotoxic T cells, suppressing the immune response[177,178]. Such mechanisms are similar and have already been described for HCC. Despite this, not much literature is present on TILs in iCCA, focusing mainly on immunohistochemistry analysis in terms of number and presence of CD4+ and CD8+ cells.
Briefly, CD4+ cells have been found to infiltrate tumor specimens. On the contrary, CD8+ cells are present at the tumor margin, correlating with prognosis. Patients with iCCA with low infiltration of cytotoxic T cells show a worse prognosis[179,180]. The presence of both TIL and PDL-1/PD-1 expression makes iCCA possibly suitable for immunological target therapies[181,182].
NK cells in iCCA
Less efforts have been made to unveil the specific role of NK cells in iCCA[151]. However, several preclinical and clinical studies have assessed the activity of NK cells against iCCA. The use ofin vitrocytokine-activated NK cells in combination with cetuximab, the mAb against EGFR, has shown benefits in a higher antibodydependent cellular cytotoxicity response against human iCCA cell lines such as HuCCT-1 and OZ[183]. Moreover, the multiple infusions ofex vivo-expanded human NK cells into iCCA xenograft mice (HuCCT-1 tumor-bearing nude mice) resulted in NK cell-mediated cytolytic response with inhibition of tumor growth[184].
Recently, an elevated intra-tumoral expression of CXCL9, an IFN-γ inducible chemokine, was associated with a large number of tumor-infiltrating NK cells, leading to favorable postoperative survival in patients with iCCA[185]. Additionally, elevated expression of NKG2D ligands in human iCCA correlate with improved DFS and OS in patients[186]. Although these findings hold promise, further studies are needed to investigate the role of NK cells in the pathogenesis of iCCA. In fact, similar to HCC, strategies with the aim of evading NK cell immunosurveillance in CCA have been reported. For instance, iCCA cells are able to induce apoptosis in NK cells, via the Fas/FasL pathway, and escape the inflammatory response by upregulating the antiapoptotic c-FLIP system[187]. On the other hand, several nucleotide polymorphisms (SNPs) located within the NKG2D receptor gene (KLRK1) have been linked to impaired NK cell effector functions and higher risk of cancer[188].
Specifically, the development of CCA in patients with PSC have been associated with polymorphisms in the NKG2D gene, thus patients who are homozygous for the NKG2D alleles are likely to develop CCA. These data clearly support different roles and clinical impacts of NK cells in iCCA disease. However, it is still not clear how these activities are related to the specific blood circulating and liver resident NK cells.
FUTURE CHALLENGES
The recent advances in the understanding the important cross-talk between cancer cells and cell infiltrating TME allowed to identify various mechanisms underlying tumor development and progression. The pathways beyond this cells-cells cooperation have been demonstrated to have harmful role in impaired immune cells activation and also in therapeutic response. In particular, NK cells have been reported to have a prominent role in maintaining the homeostasis in the liver even in case of liver tumors. Yet, new therapies based on targeting NK cells with the aim to restore their impaired cytotoxic activity within tumor are gaining attention. In the era of precision medicine, this challenging research area could open the possibility to develop new potential therapeutic strategies in combination with conventional therapies for the treatment of HCC and iCCA patients.
CONCLUSION
In this review, we have examined the key pathways underlying TME cell-cell communications, with deeper focus on the role of natural killer cells in primary liver tumors, such as HCC and iCCA, as new opportunities for immune-based therapeutic strategies.
ACKNOWLEDGEMENTS
The authors thank Dr. Soldani C, Dr. Franceschini B and Dr. Costa G from the Hepatobiliary Immunopathology Laboratory, Humanitas Clinical and Research Center – IRCCS, Rozzano, Milan (Italy) for their contribution in the reviewing the pertinent literature.
 World Journal of Gastroenterology2020年33期
World Journal of Gastroenterology2020年33期
- World Journal of Gastroenterology的其它文章
- Tumor necrosis factor alpha receptor 1 deficiency in hepatocytes does not protect from non-alcoholic steatohepatitis, but attenuates insulin resistance in mice
- Resveratrol alleviates intestinal mucosal barrier dysfunction in dextran sulfate sodium-induced colitis mice by enhancing autophagy
- Acute liver failure and death predictors in patients with dengue-induced severe hepatitis
- Surveilling Russell body Helicobacter pylori-negative gastritis: A case report and review of literature
- Exploring the food-gut axis in immunotherapy response of cancer patients
- Liver fat accumulation measured by high-speed T2-corrected multi-echo magnetic resonance spectroscopy can predict risk of cholelithiasis