
A sulfotransferase gene BnSOT-like1 has a minor genetic effect on seed glucosinolate content in Brassica napus
2020-10-21 10:02:36YangmingWangShubeiWanHaoFanMaoYangWeiyanLiRongzhanGuan
The Crop Journal 2020年5期
Yangming Wang,Shubei Wan,Hao Fan,Mao Yang,Weiyan Li,Rongzhan Guan,*
aState Key Laboratory of Crop Genetics and Germplasm Enhancement,Nanjing Agricultural University,Nanjing 210095,Jiangsu,China
bJiangsu Collaborative Innovation Center for Modern Crop Production,Nanjing Agricultural University,Nanjing 210095,Jiangsu,China
ABSTRACT
Keywords:
1.Introduction
Glucosinolates(GSLs)are a group of secondary metabolites present in thousands of plant species.GSL degradation products may function as antimicrobial compounds[1,2],deterrents of generalist herbivores[3],herbicides[4],and anti-carcinogenic agents[5,6],but their toxicity to animals restricts their utility in the protein-rich seed meal obtained from Brassica oilseed species[7].More than 120 different GSLs have been identified.Each GSL consists of a glucose group,a sulfonated oxime group,and an amino acid-derived R-side chain.On the basis of the source of the R-side chain,GSLs can be divided into aliphatic,indolic,and aromatic GSLs,which are derived from methionine,tryptophan,and phenylalanine or tyrosine,respectively[8].The biosynthesis of GSLs has been largely elucidated in plant species such as Arabidopsis thaliana[9,10].The process consists of three phases:amino acid sidechain elongation,core structure synthesis,and side-chain modification[11].The wide variety of known amino acid sidechain elongations and secondary modifications,including hydroxylations[12],O-methylations[13],desaturations[14],glycosylations[15],and acylations[16],results in the structural diversity of GSLs[17].
In A.thaliana,many GSL-associated genes have been identified,including 58 GSL biosynthesis genes,3 GSL transport genes,and 17 GSL degradation genes[18].Thus,the genetic network regulating the GSL content in A.thaliana appears to be relatively small.However,a search of four published Brassica genomes based on a combination of syntenic and nonsyntenic homology analyses with A.thaliana GSL-associated gene sequences as queries revealed substantially more GSL-related genes in Brassica species than in A.thaliana,likely because of the genomic expansion that occurred during the evolution of Brassica species[18].Most of these Brassica annotated GSL-associated genes have not been thoroughly characterized,and probably more GSL biosynthesis-associated genes remain to be discovered.Three regulatory genes associated with GSL content have been identified on chromosomes A09,C02,and,C09 as major QTL via linkage mapping and association mapping.These genes include homologous regulatory genes(BnHAG1)which are MYB28 transcription factor genes.They regulate the methionine side-chain elongation phase and core structure synthesis phase of GSL biosynthesis.It has been shown[19–23]that the MYB28 up-regulates the expression of certain genes(e.g.MAM1–3,CYP79/83,SUR1,and ST5b/c)involved in the biosynthesis of aliphatic GSLs,which comprise major GSLs of B.napus seeds.Loss-offunction mutations of the three BnHAG1 genes in Brassica napus led to low total seed GSL content.So-called low seed GSL content means that the seed GSL content in a cultivar is less than 40μmol g-1,but not zero.Among cultivars with low GSL content,there exists seed GSL content variation which is controlled by polygenes[24–29].In some QTL studies,at least eight unstable QTL with minor effects have been unstably detected.To date,no minor genes influencing GSL content have been functionally studied.
Sulfotransferase(SOT)genes are involved in GSL biosynthesis[30].SOT family members have been detected in most studied organisms[31].The family includes many members with seven conserved domains,of which three(PF00685,PF03567,and PF13469)are present in most SOT proteins[32].Several SOTs catalyze the sulfonation of diverse compounds[33],including flavonol[34],brassinosteroids[35],salicylic acid[36],tyrosylprotein[37],gibberellic acids[31],jasmonic acids[38],and GSL[39].The sulfonation of bioactive molecules affects plant growth and development[40],adaptation to stress[41],pathogen resistance[36],and detoxification[42].A cluster analysis of 17 A.thaliana SOTs and 35 Oryza sativa SOTs divided SOT genes into seven subfamilies[33].SOTs within the same subfamily or with highly similar sequences may have diverse functions because of differences in substrate specificity[31].Accordingly,SOT gene functions cannot be simply inferred by sequence alignment,and careful study is required for the comprehensive functional annotation of the numerous SOT genes.There are three AtST5 SOT genes in A.thaliana(AtST5a,AtST5b,and AtST5c)[30].The enzymes encoded by these genes convert desulfurized GSLs into GSLs with a specific core structure.In B.napus,BnSOT16,BnSOT17,and BnSOT18 are homologs of A.thaliana AtST5a,AtST5b,and AtST5c,respectively[32].These A.thaliana and B.napus genes function similarly.However,whether other SOT genes contribute to the final enzyme-catalyzed step of GSL formation is unclear.
In classical quantitative genetics,the theory and associated breeding methods were developed based on the multifactorial or polygene hypothesis,and have sustained the genetic improvement of animals and plants[43].In modern quantitative genetics,the approaches used for breeding and quantitative trait locus(QTL)research have been established based on the“major gene plus polygene”genetic system theory[44,45].Although to date,many major QTL have been mapped and verified,relatively little is known about the minor genes influencing quantitative traits[46].
The present study is an investigation of the role of polygenes,using a glucosinolate-associated gene of minor effect in the natural biological state as an example.The gene BnaA09g53490D was occasionally identified in functional analysis experiments of plant height candidate genes in B.napus.To clarify the function of the gene,genetic transformation and biochemical assays associated to GSL content were conducted together with transcriptomic determination experiments.Furthermore,QTL mapping was performed to statistically evaluate the genetic effect of the gene and to examine whether it is a major or minor gene.
2.Materials and methods
2.1.Plant materials
The accession M176 with low erucic acid content and low glucosinolate content in seeds(double-low)and double-low cultivar Zhongshuang 11(ZS11)were used as parents and for genetic transformation.A backcross population,BC1[ZS11×(ZS11×M176)],was generated and evaluated to map glucosinolate QTL as described in a previous study of plant height QTL[47].The seed GSL contents of the two parents differed by about 10μmol g-1dry meal.
2.2.GSL content assay
The seed GSL contents of 150 plants in the BC1population were determined with the VECTOR 22/N Fourier transform infrared spectrometer(Bruker corporation,Karlsruhe,Germany).The seed GSL contents of the transgenic lines and the parents(ZS11 and M176)were measured by highperformance liquid chromatography(HPLC)at the Laboratory of Quality & Safety Risk Assessment for Oilseed Products(Wuhan),Ministry of Agriculture,Oil Crops Research Institute,Chinese Academy of Agricultural Sciences[21].Analyses were performed with three biological replicates.
2.3.Quantitative trait locus mapping
The BC1population was genotyped using the Brassica 60 K Infinium Bead Chip Array(Illumina,San Diego,CA,USA)and genome-wide single-nucleotide polymorphism(SNP)genotypes were obtained as described previously[47].Inclusive composite interval mapping(ICIM)was used for QTL detection as described previously[48,49].The LOD threshold for QTL detection was determined by permutation test with 1000 iterations and a significance level of 0.05.
2.4.RNA preparation
Total RNA was extracted from tissues with the TRIzol reagent(Invitrogen,Carlsbad,CA,USA)and then digested with RNasefree DNase I(Takara,Dalian,China)to eliminate any residual genomic DNA.RNA quality and concentration were determined by electrophoresis and spectrophotometry(One Drop OD 1000+,Shanghai,China).The RNA was used as template to synthesize cDNA with the PrimeScript II 1st Strand cDNA Synthesis Kit(Takara).
2.5.Gene cloning
Allelic sulfotransferase genes for BnaA09g53490D were cloned from ZS11 and M176 and named Bnsot-like1 and BnSOT-like1,respectively.The full-length BnaA09g53490D open reading frame was amplified by polymerase chain reaction(PCR)from cDNA produced from ZS11 and M176,using the gene-specific primers BnaA09g53490D-cds-F(5′-ATGACATCATCATCTGTTCCTA-3′)and BnaA09g53490D-cds-R(5′-TCAACAAGAAAATTTCAGACC-3′).The PCR program was as follows:94 °C for 5 min;35 cycles of 94 °C for 30 s,58 °C for 30 s,and 72 °C for 60 s;72 °C for 5 min.The amplified fragment was inserted into the pEASY-Blunt Cloning Kit vector(TransGen,Beijing,China)and sequenced to confirm accuracy.
2.6.Sequence analysis
DNA sequences of the B.napus sulfotransferase(SOT)gene family were retrieved from the Genomic Variation Database of B.napus(http://rapeseed.biocloud.net/home).Clustal X[50]was used for multiple sequence alignment.A phylogenetic tree was constructed with MEGA 5.0[51]by the neighborjoining method,with a bootstrap test with 1000 replications.Predicted A.thaliana SOT amino acid sequences were retrieved from TAIR(https://www.arabidopsis.org/index.jsp)by search the A.thaliana gene ID in[31].
2.7.Gene expression analysis
Total RNA was extracted from B.napus roots,stems,leaves,flowers,buds,siliques,and cotyledons for semi-quantitative PCR,with the constitutively expressed Actin7 used as the reference control.The reference gene-specific primers were BnACTIN7-qpcr-R (5′-ATTCAGCCCCTTGTTTGTG-3′) and BnACTIN7-qpcr-R(5′-GTAAGCGTCTTTTTGACCCAT-3′).The target gene-specific primers were BnaA09g53490D-qpcr-F(5′-AGTGAGATTGTGAAG TTGTGT-3′)and BnaA09g53490D-qpcr-R(5′-AAGGACTCACTCAAAGTATCTC-3′).The PCR program was as follows:94 °C for 5 min;30 cycles of 94 °C for 30 s,60 °C for 30 s,and 72 °C for 30 s;72 °C for 5 min.
2.8.Transformation with BnSOT-like1
Bnsot-like1 and BnSOT-like1 were incorporated into the pBI121 vector to generate the pBI121::BnSOT-like1 and pBI121::Bnsotlike1 recombinant plasmids for gene expression under the control of the CaMV 35S promoter.The genes were inserted into ZS11 plants by the floral dip method[52].Three independent T3homozygous transgenic lines were randomly selected for further analysis.
2.9.RNA sequencing
Plants overexpressing BnSOT-like1(OE-M)were evaluated by RNA sequencing,with wild type ZS11 plants serving as the control.Seedlings were grown in pots containing peat:vermiculite(1:1 v/v).Leaves of 8-week-old plants were harvested,immediately frozen in liquid nitrogen,and stored at-80 °C.Total RNA was extracted from the leaves as described above.The RNA was used to construct sequencing libraries that were sequenced with the HiSeq 2500 platform(Illumina).Clean reads were mapped to the B.napus reference genome Darmor v4.1(http://www.genoscope.cns.fr/brassicanapus/data/) with the default parameters of TopHat2[53].The FPKM(fragments per kilobase of exon per million mapped reads)value of each gene was calculated with Cuffdiff[54].Differentially expressed genes(DEGs)(|log2(Fold change)|≥1,false discovery rate<0.05)were identified with DESeq2[55].
3.Results
3.1.Positional cloning
The sulfotransferase gene locus BnaA09g53490D was identified in a fine mapping interval for locus Bndwf1 influencing plant height and architecture[47].The two alleles were cloned from the two parents(M176 and ZS11)from the mapping interval with the same pair of primers,and were designated as BnSOT-like1 and Bnsot-like1,respectively.Sequencing revealed that both genes are 972 bp long and encode 323 amino acids,and differ from the homologous gene(BnaA09g53490D)annotated in GENOSCOPE(http://www.genoscope.cns.fr/brassicanapus/data/),but that all three genes contain the conserved regions I,II,and IV characteristic of SOT family genes(Fig.1).The homologous gene Bra017365 located at the same chromosomal position in the B.rapa genome(http://brassicadb.org/brad/)contains a 60-bp insertion/deletion sequence.
3.2.Sequence analysis
A phylogenetic tree of SOT genes showed that BnSOT-like1 and Bnsot-like1 are closely related to the Arabidopsis gene AtSOT12(Figs.2 and 3),although the amino acid sequence identities were only 77.9% and 76.1%,respectively.A comparison with BNSTs(steroid SOT),which sulfonates brassinolides[41],indicated that BnSOT-like1 and Bnsot-like1 are more closely related to BNST1 than to AtSOT12,with sequence identities of 85.4%and 94.1%,respectively(Figs.2,3).A previous study[35]confirmed that AtSOT12 encodes a brassinosteroid SOT involved in hormone regulation.Based on these sequence alignments,BnSOT-like1 was speculated to affect plant height,the reason that it was originally identified as a candidate gene controlling plant height.
In Brassicaceae,SOT genes involved in GSL biosynthesis include the desulfo-glucosinolate SOT genes(ds-Gl SOTs)BnSOT16-a/b/c/d,BnSOT17-a/b,and BnSOT18-a/b/c/d/e/f as well as AtSOT16,AtSOT17,and AtSOT18[32].To identify gene functions,BnSOT-like1 and Bnsot-like1 were compared with these putative ds-Gl SOT genes(Fig.S1).Protein sequence alignments revealed that BnSOT-like1 and Bnsot-like1 differed from the ds-Gl SOTs,although they all shared the conserved sequences characteristic of sulfotransferase.It thus remained unclear whether BnSOT-like1 and Bnsot-like1 contribute to GSL biosynthesis in B.napus.Some predicted SOT genes in the Brassica genome were moderately similar to BnSOT-like1 and Bnsot-like1, but their functions remained unclear.
3.3. Gene expression
BnaA09g53490D was expressed in all analyzed tissues, but most highly in the silique. The homolog was similarly expressed in the two accessions ZS11 and M176 (Fig. 4).
3.4. Overexpression of BnSOT-like1 increased seed aliphatic GSL content
Transformation of the parental cultivar ZS11 resulted in respectively two and five independent transgenic plants (Fig.5) overexpressing BnSOT-like1 and Bnsot-like1. In the corresponding homologous transgenic lines obtained by selfing,target gene expression, plant height, and seed GSL composition were measured. The SOT genes were expressed in leaves much more highly than in the wild type ZS11 (Fig. 6). The plant architecture of the transgenic lines was not markedly altered (Table S1). This finding suggested that BnSOT-like1 did not act as a sulfotransferase that sulfonates brassinolides,although it was more similar to BnSTs that sulfonate brassinolides, than were many other SOT genes in B. napus,given that sulfonation deactivates brassinolides and compresses plant architecture [41].
The seed total GSL content of BnSOT-like1 was 7.33 times higher than that of the ZS11. In contrast, the seed GSL contents of plants overexpressing the Bnsot-like1 gene were not sharply different from that of ZS11 (Table 1, Fig. S2). These findings indicated that BnSOT-like1 was involved in GSL biosynthesis, that its overexpression increased seed GSL content, and that the Bnsot-like1 allele did not increase seed GSL content.
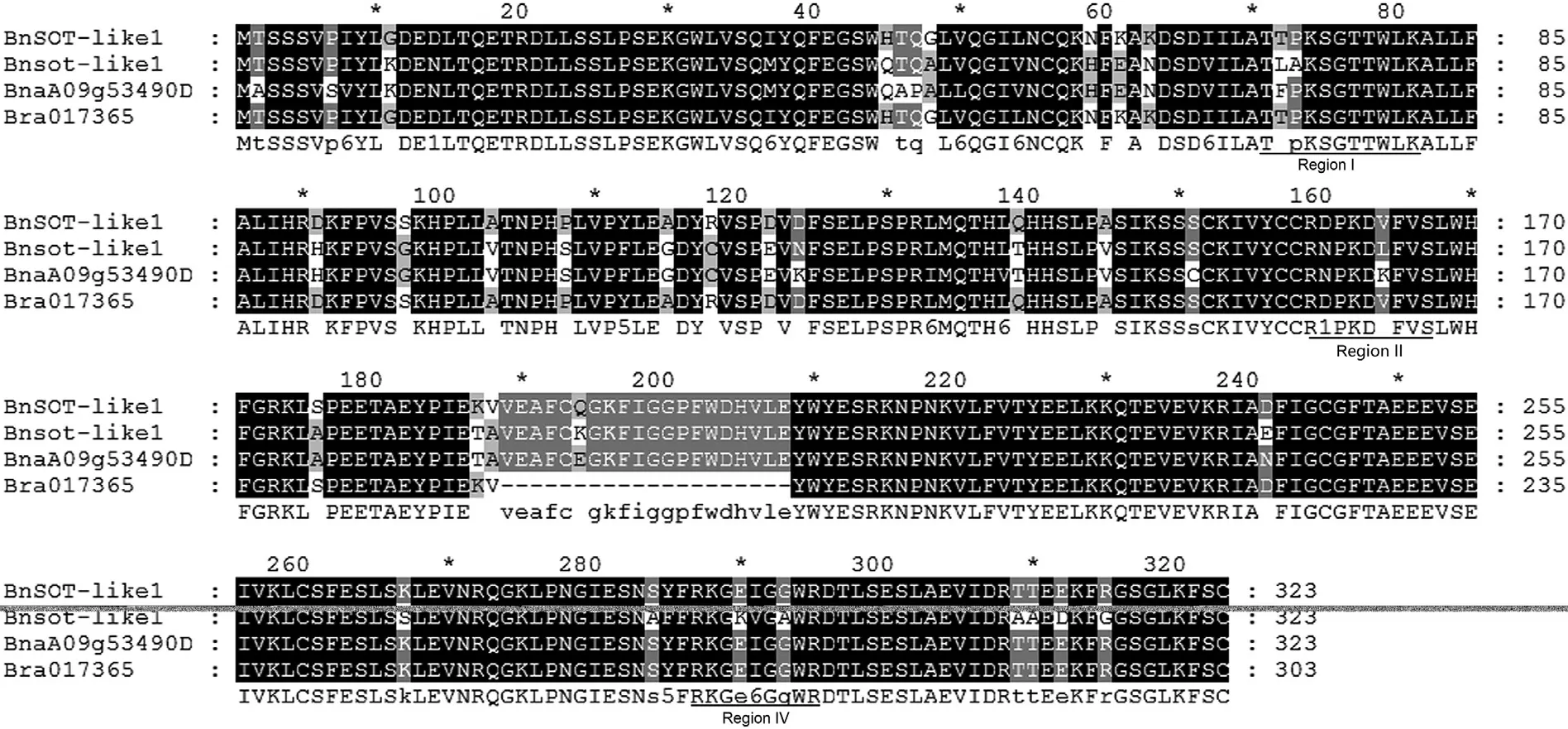
Fig.1–Multiple sequence alignment of Bnsot-like1,BnSOT-like1,BnaA09g53490D,and Bra017365.The positions of conserved domains(I,II,and IV)are underlined.
Table 1 shows that the seeds of transgenic plants overexpressing BnSOT-like1 contained much more of the major aliphatic GSL components,including desulfoprogoitrin and desulfogluconapin,than the wild type.This finding suggests that BnSOT-like1 catalyzes the formation of the aliphatic GSL core structure but does not affect the elongation of the GSL side chain.The protein encoded by Bnsot-like1 did not catalyze GSL formation because of a sequence alteration.Thus,sequence mutations in the conserved functional regions resulted in altered gene functions,with BnSOT-like1 encoding a SOT enzyme that uses aliphatic desulfurized GSLs as substrates,whereas Bnsot-like1 encodes a nonfunctional SOT enzyme.
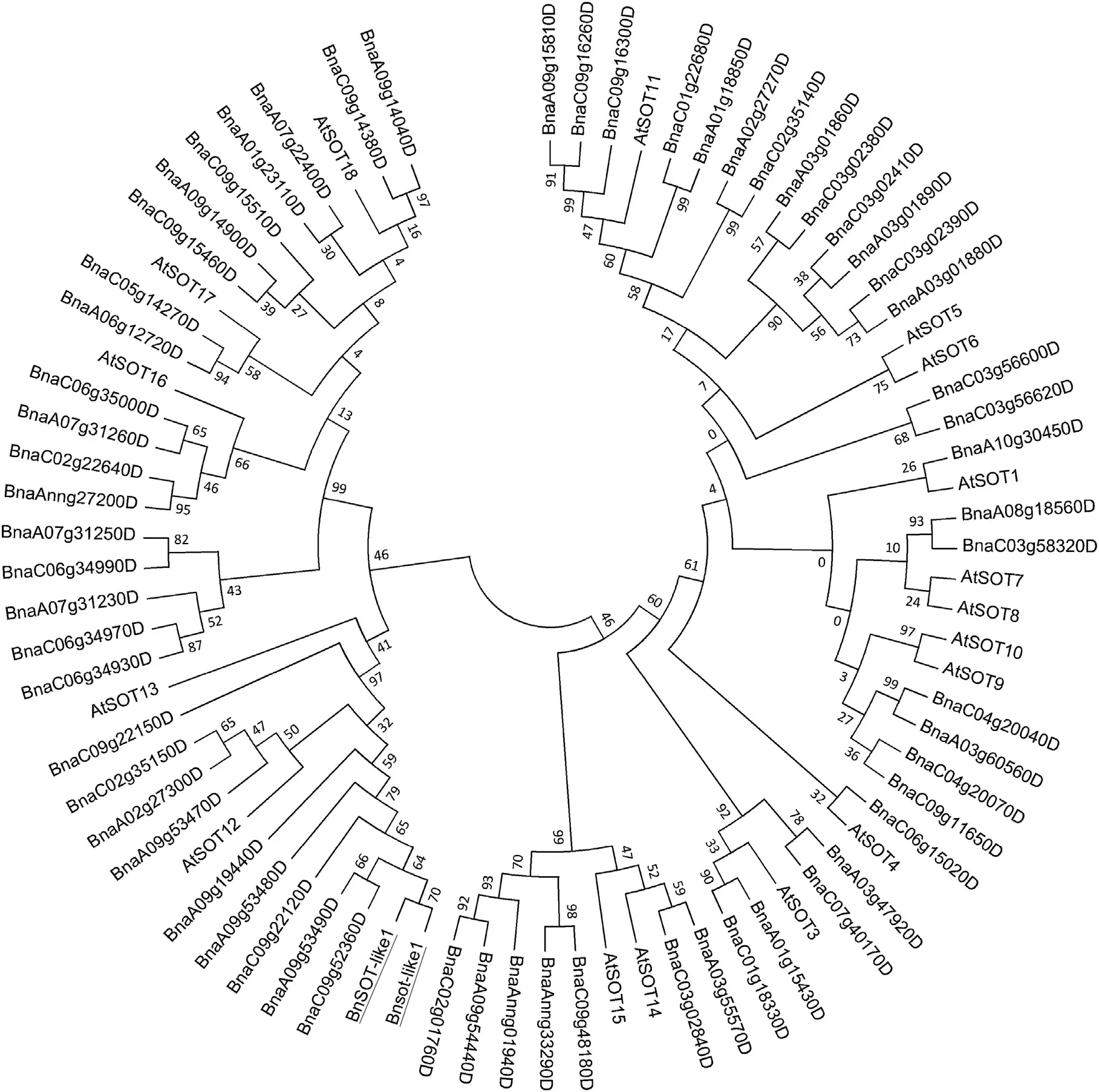
Fig.2–Phylogenetic tree of SOT proteins in B.napus and A.thaliana.
3.5.BnSOT-like1 is a minor gene affecting seed GSL content
ICIM with a LOD threshold of 3.12 detected only one putative QTL(qGSLC02)for seed GSL content(Fig.7,Table 2).This QTL(LOD=4.11)was located at 8 cM on the C02 chromosome and explained 11.1%of the phenotypic variance.
However,the QTL qGSLC02 was not associated with the observed SOT functions.A reasonable explanation for this finding is that BnSOT-like1 is a minor gene in its natural biological state and the effects of the BnSOT-like1 alleles are too small to be detected.To support this inference,we carefully examined the LOD profile across the whole genome(Fig.7),and found at the physical position of the BnSOT-like1 a small peak with LOD score 0.48,far below the LOD threshold for QTL detection(Fig.7,Table 2).This finding suggested that the BnSOT-like1 allele acted as a minor QTL with small effects undetectable under standard strict criteria,contributing a small proportion(1.1%)of phenotypic variance in the population.
There were many other small peaks below the threshold in the LOD profile.These could correspond to numerous undetected QTL for seed GSL content.It may be concluded that seed GSL content is controlled by major genes plus polygenes,as suggested in previous reports[18,26,28,56–59].

Fig.3–Multiple sequence alignment of Bnsot-like1,BnSOT-like1,BNSTs,and AtSOT12.The positions of conserved domains(I,II,and IV)are underlined.
3.6.RNA sequencing analysis
A total of 10,195 differentially expressed genes(DEGs)were identified in BnSOT-like1 transformants and wild-type plants,of which respectively 4621 and 5574 genes were up-and down-regulated.These DEGs included 12 genes involved in the aliphatic GSL biosynthesis pathway,all of which showed up-regulated(approximately 2.00–4.95-fold)expression(Table 3).However,there were no differences in the expression of regulator genes(the three BnHAG1 transcription factor genes).These results suggested that overexpression of BnSOT-like1 up-regulated the expression of genes involved in the aliphatic GSL biosynthesis pathway,thereby increasing seed GLS content.
4.Discussion
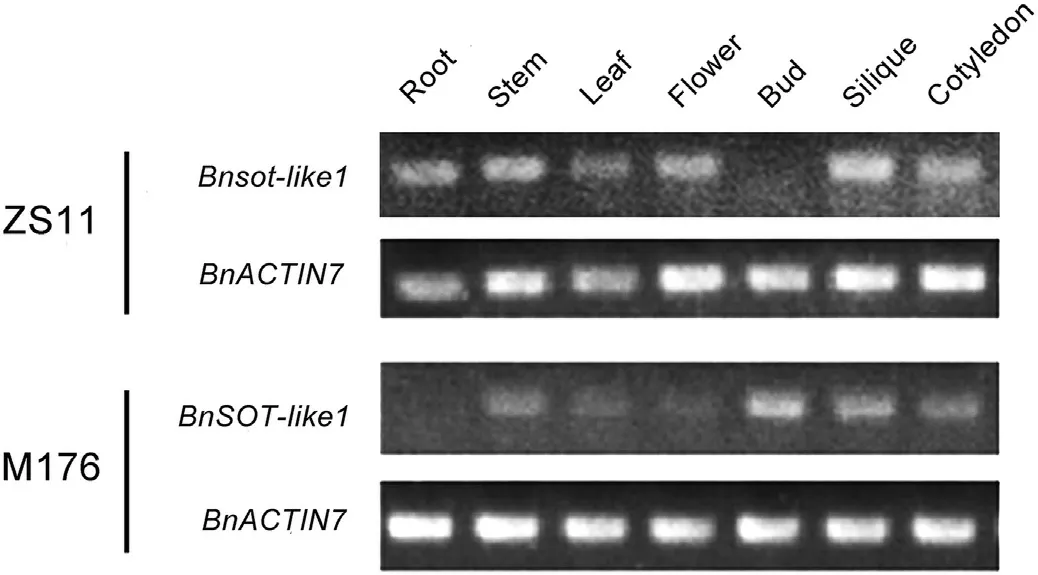
Fig.4–Expression pattern of BnSOT-like1 and Bnsot-like1(BnaA09g53490D homologs)in seven tissues of M176 and ZS11 by semi-quantitative RT-PCR.
In addition to the three reported major genes(BnHAG1)controlling seed GSL content[18],we have identified a minor gene BnSOT-like1 which has a small genetic effect on seed GSL content in B.napus.In quantitative genetics,genes with large effects are called major genes,whereas genes with undetectable effects are considered to be polygenes or minor genes.Theoretically,mutations of GSL biosynthesis pathway genes will affect GSL contents.However,every gene in the allotetraploid species B.napus has at least three copies,resulting in gene function redundancy.In this case,any loss-or gain-offunction mutation to a biosynthetic gene will be compensated by the other genes with the same function.Thus,the mutated gene will not markedly influence the plant phenotype and the biosynthetic genes in polyploid species are inherited as minor genes,which may be undetectable.There is relatively little information about minor genes,and it is unclear why a single minor gene contributes minimally to phenotypic variations.Statistical geneticists attempting to explain this phenomenon have suggested that the small effects of minor genes,which are subtle and undetectable by common QTL mapping approaches,are due to the large numbers of gene influencing a trait.However,for molecular biologists,every verified gene has significant effects,with a clear mechanism underlying gene function in biochemical and molecular processes,and minor genes for quantitative trait variations may be detected in natural populations.Thus,the contributions of minor genes to biomolecular networks should be elucidated further.
Fig.5–Representative screening of transgenic plants by PCR.PC,pBI121::BnSOT-like1 vector was used as positive control;NC,empty control(DNA from ZS11 without vector)as negative control;M,marker DL2000.
In B.napus,a total of 96 SOT genes were identified via B.napus database searches.These genes are distributed on all chromosomes(Fig.S3),but many of them have not been functionally characterized.The present study identified a sulfotransferase gene(BnSOT-like1)involved in GSL biosynthesis.There are probably more SOT genes involved the formation of the GSL core structure than expected in B.napus,and there are probably more kinds of genes with minor effects in GSL biosynthetic pathway influencing GSL content in natural populations.Our QTL study suggested the presence of unidentified QTL for GSL content,and are in accord with the polygene hypothesis.
In this study,the SOT gene involved in GSL biosynthesis tends to be expressed in siliques,affects seed GSL content,and is a minor gene.Overexpression of the SOT gene leads to elevation of seed GSL content.Undoubtedly,gene expression in GSL biosynthesis is affected by biotic and abiotic factors.Thus,environmental and biological influences on polygenes may affect GSL content.This phenomenon is also in agreement with the multifactor hypothesis of quantitative trait expression.
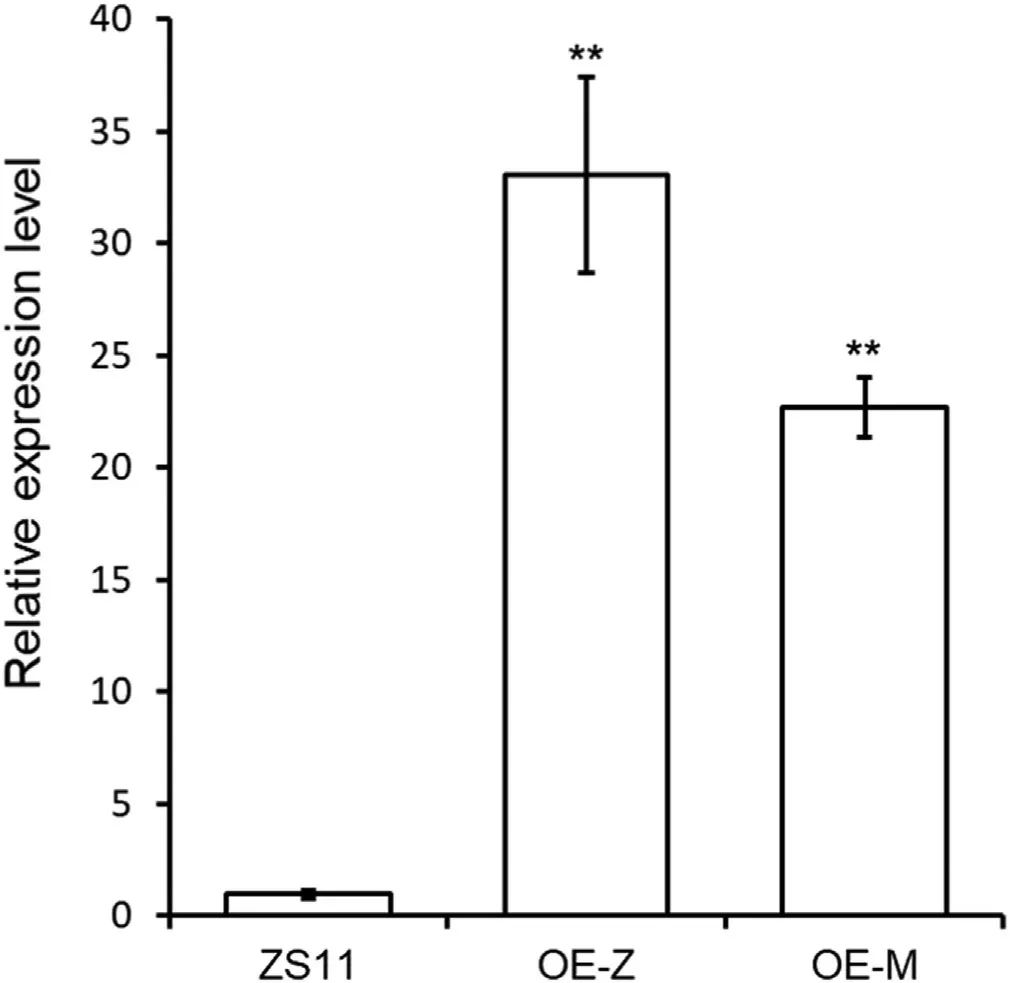
Fig.6–Expression levels of BnSOT-like1 in ZS11,OE-Z,and OE-M.OE-Z and OE-M denote mean levels in transgenic lines overexpressing Bnsot-like1 and BnSOT-like1,respectively.**denotes significant difference at the 0.01 probability level.
Although the SOT gene in the present study was not detected under strict QTL detection criteria because of its relatively minor genetic effect,its role may be clarified by gene overexpression experiments involving transgenic plants.This might permit the deployment of a functional polygene by genetic engineering,molecular breeding,and genome editing.
In plant and animal genetic improvement,minor genes are important.Because there are numerous minor genes affecting the phenotypes of quantitative traits,the aggregation of numerous minor genes may lead large effects as measured by indicators such as combining ability.Accumulation of favorable minor genes,as indicated by general combining ability increase,have promoted breeding advances for more than a hundred years[60].Undoubtedly,polygene aggregation based on modern or classical quantitative genetics is effective.In modern genetics and breeding,selection based on marker-associated QTL is more reliable than selection in conventional breeding.Mining QTL is a practical objective.
Minor genes are difficult to detect statistically owing to the small gene effect.However,there is still a possibility to detect minor genes or QTL by adjusting the mapping strategy.First,lowering detection criteria of QTL may reveal many more QTL including minor QTL.Second,QTL with minor effect may be detected in only a single environment.If these unstably detected QTL are not ignored,the probability of discovering minor QTL will increase.Third,use of structurally simplified populations such as chromosome segment substitution lines may facilitate minor QTL detection[61,62].Finally,morepowerful multi-locus approaches for QTL mapping, such as RTM-GWAS [63,64], may reveal more QTL with small effects.
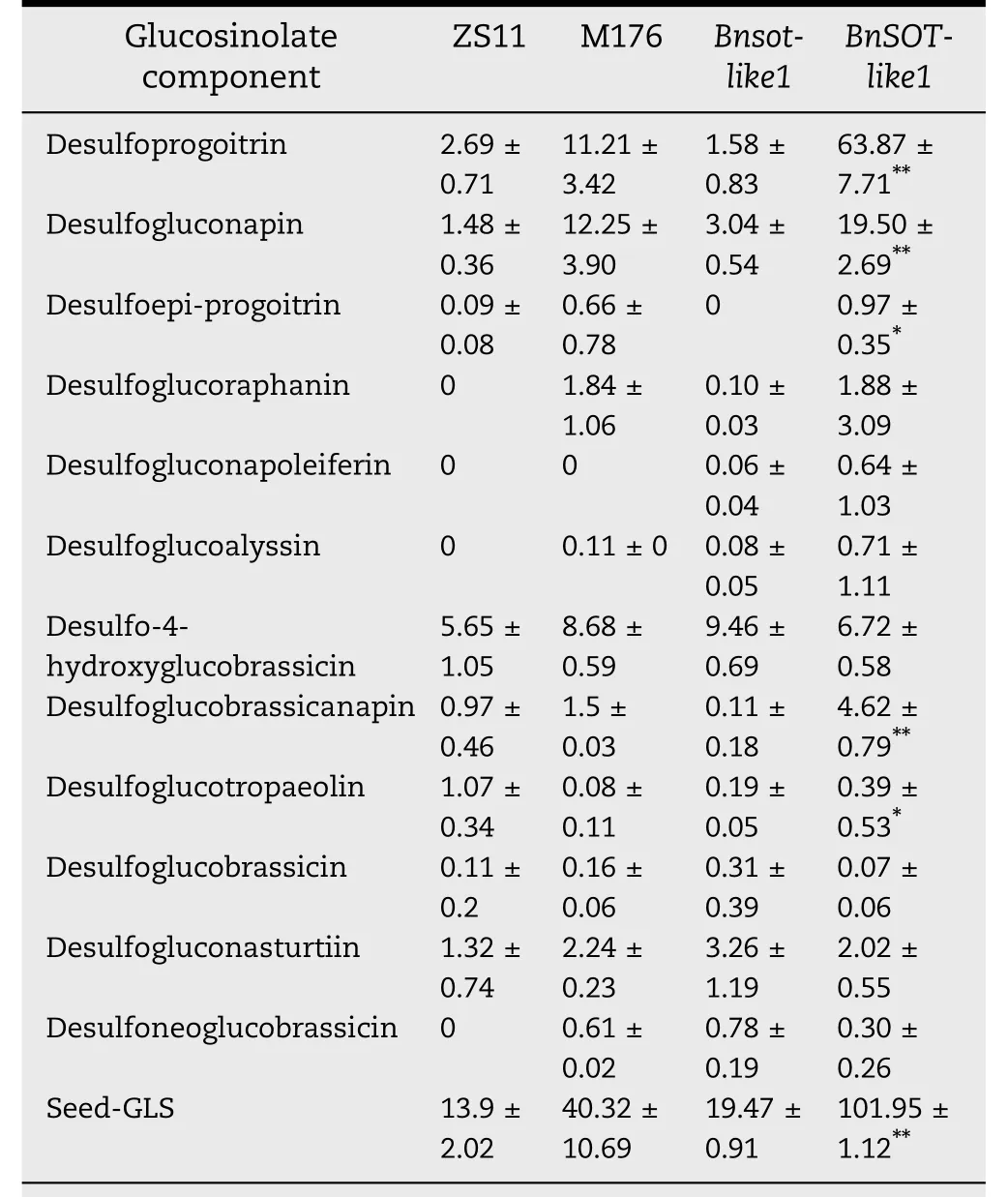
Table 1–Average content of seed GSL component in transgenic lines.
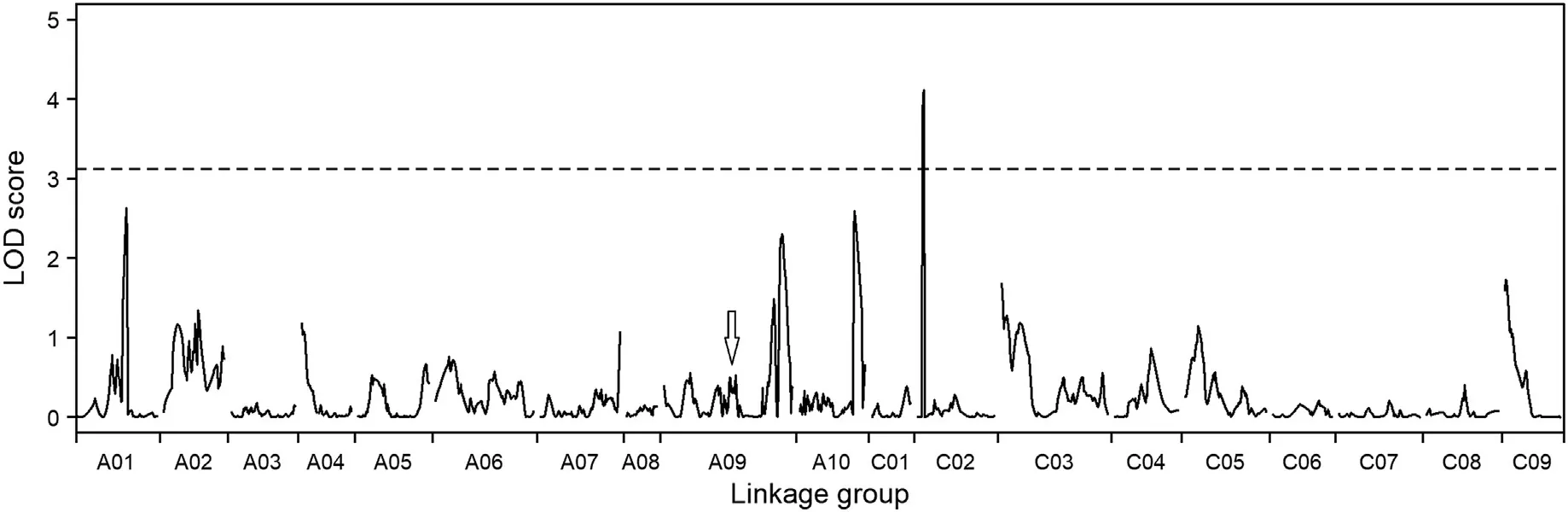
Fig.7–LOD score profile of QTL mapping for seed GSL content on 19 chromosomes including that of A and C sub-genomes in B.napus.The broken horizontal line indicates the LOD threshold of 3.12.The arrow indicates the location of BnSOT-like1.One region on chromosome C02 reached the maximum estimated LOD score of 4.11.
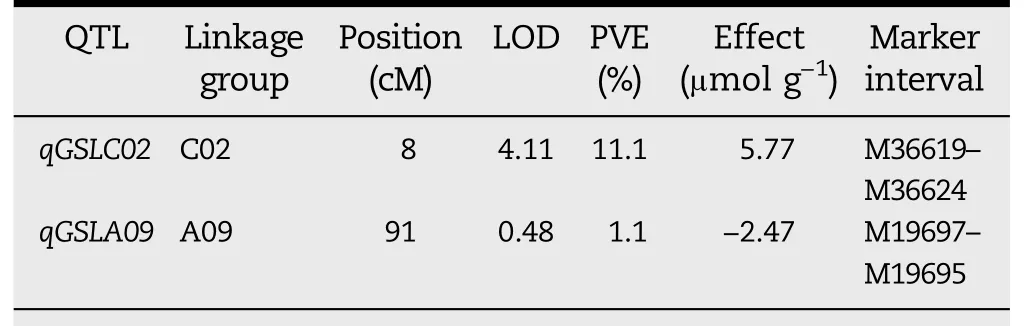
Table 2–QTL mapping of seed total glucosinolate content by ICIM.
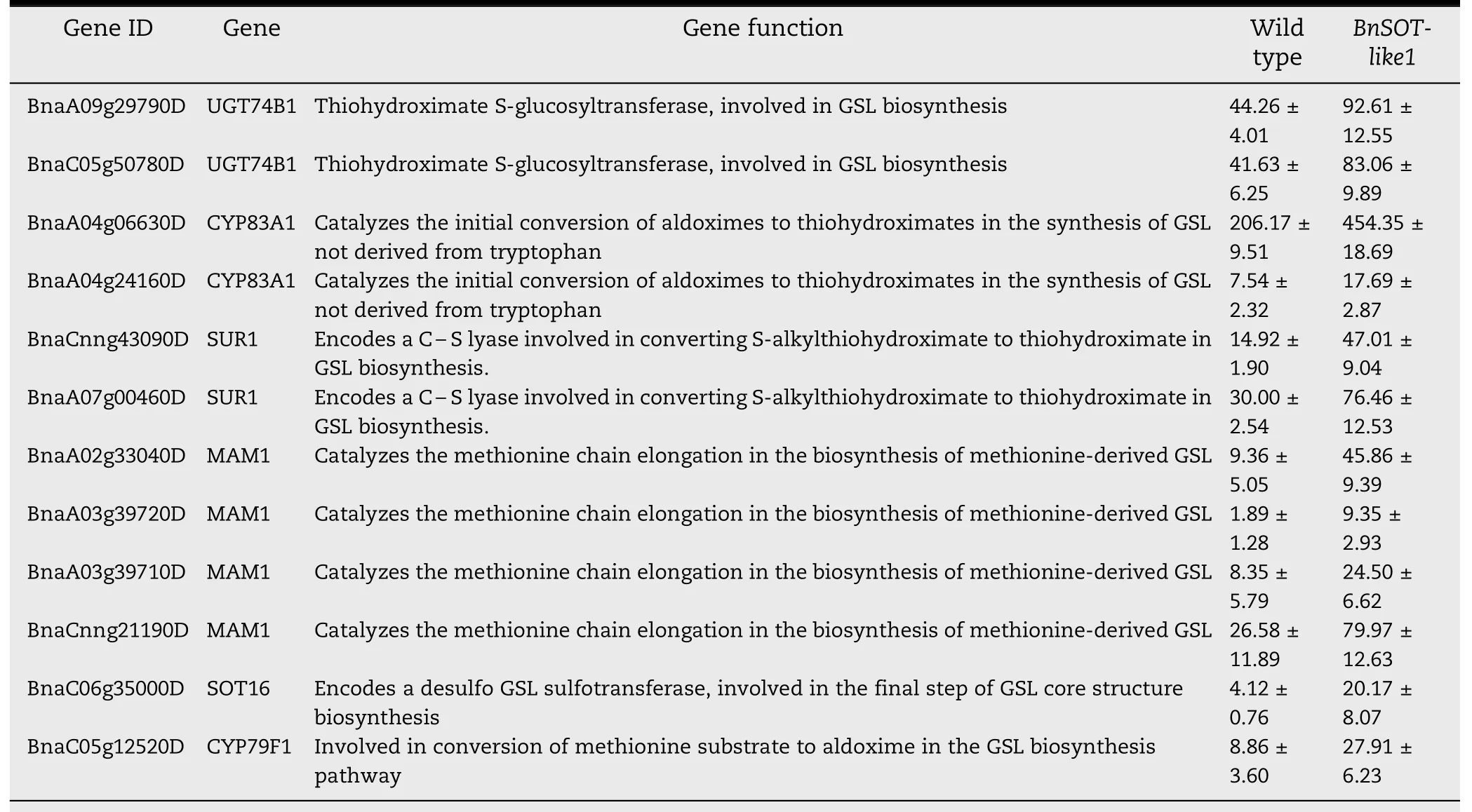
Table 3–Differentially expressed genes of GSL biosynthesis pathway in leaves.
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2020.07.003.
Declaration of competing interest
Authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the National Key Research and Development Program of China(2018YFD0100600),the National Natural Science Foundation of China(31270386),and the Cyrus Tang Seed Innovation Center at Nanjing Agricultural University.
Author contributions
Rongzhan Guan conceived and designed the study.Yangming Wang performed most of the experiments.Yangming Wang and Shubei Wan wrote the manuscript.Rongzhan Guan advised on the experiments and modified the manuscript.Shubei Wan,Hao Fan,Mao Yang,and Weiyan Li took part in the DNA extraction and QTL mapping experiments.All authors read and approved the final manuscript.

- The Crop Journal的其它文章
- Quantitative genetic studies with applications in plant breeding in the omics era
- Brief Guide for Authors
- Use of family structure information in interaction with environments for leveraging genomic prediction models
- META-R:A software to analyze data from multi-environment plant breeding trials
- Modeling and simulation of recurrent phenotypic and genomic selections in plant breeding under the presence of epistasis
- Genome-wide prediction in a hybrid maize population adapted to Northwest China