
反式聚环戊烯橡胶合成及结构分析
2020-10-11 01:28:34贺小进邱迎昕曹达鹏陈建军卜少华罗俊杰
北京化工大学学报(自然科学版) 2020年4期
贺小进 邱迎昕 王 雪 曹达鹏 陈建军 卜少华 罗俊杰
(1.中国石化 北京化工研究院燕山分院, 北京 102500; 2.中国石化 橡塑新型材料合成国家工程研究中心, 北京 102500;3.北京化工大学 化学工程学院, 北京 100029)
引 言
反式聚环戊烯橡胶(TPR)是以环戊烯为单体,在齐格勒-纳塔催化剂作用下通过开环聚合制得,其生胶强度大,加工性能好,拉伸后可结晶,硫化胶回弹性及耐磨性优异,生热低,滚动阻力小,非常适合用于制作载重车轮胎及绿色节能轮胎。
20世纪70年代国外曾开展过TPR的合成研究,建成了中试装置,早期采用钼盐或钨盐- 烷基铝二元催化体系,后采用活性较高、稳定性较好的三元催化体系。国内庞德仁等[1-3]和宋志华等[4]也曾开展过实验室研究,所用催化剂为钨盐- 烷基铝- 活化剂三元催化体系,聚合物反式链节结构含量约90%,得到的TPR性能接近天然橡胶(NR)[1-3]。
环戊烯可从裂解石油制乙烯的副产碳五馏分中制得,早期由于聚合单体环戊烯的短缺致使TPR发展受到很大限制,近年来随着碳五资源的丰富,碳五包括环戊二烯及环戊烯的有效利用引起各方重视。2018年我国石油裂解制乙烯副产碳五量约为300万吨。碳五馏分中含15%~20%的环戊二烯,约3%的环戊烯,高附加值TPR的开发重新引起人们的关注[5-9]。日本瑞翁公司在环戊烯聚合研究方面做了大量工作,主要涉及环戊烯的均聚以及与含苯环化合物或与降冰片烯的共聚,并在链末端引入烷氧基硅烷,得到高耐磨、低生热、低滚阻的新型橡胶材料,并改善了其抗湿滑性[10-16]。为配合中国石化碳五资源的综合利用,本文开展了TPR的合成研究,确定了聚合催化剂制备及TPR合成工艺条件,制备的催化剂活性及稳定性较文献值[3,10]有一定提高;并对TPR的结构及性能进行了初步分析,得到的TPR可满足通用橡胶的要求。
1 实验部分
1.1 实验原料
环戊烯,试剂级,纯度大于98%,阿拉丁化学品有限公司;甲苯,分析纯,北京化工厂;正己烷,工业级,中国石化北京燕山分公司合成橡胶厂。以上试剂使用前均经5A分子筛浸泡至含水质量分数小于10×10-6。WCl6,试剂级,三异丁基铝,1.1 mol/L甲苯溶液,阿拉丁化学品有限公司;2,6-二叔丁基-4-甲基苯酚,工业级,中国石化北京燕山分公司合成橡胶厂;高纯氮气,纯度大于99.999%,北京普莱克斯公司。
1.2 TPR的制备
向经氮气充分抽排的5 L反应釜中加入一定量的溶剂正己烷、环戊烯、WCl6溶液、四氯苯酚溶液或陈化后的催化剂溶液及分子量调节剂正丁烯,打开反应釜搅拌及低温冷浴,冷却至0 ℃左右缓慢加入三异丁基铝溶液,反应2~3 h后加入2,6-二叔丁基-4-甲基苯酚的乙醇溶液终止聚合反应。聚合物溶液用乙醇凝聚后在通风橱中除去大部分溶剂,再于60 ℃真空烘箱中干燥至恒重,之后测定聚合物结构。
1.3 分析方法
TPR相对分子质量及其分布采用LC- 10AVP型凝胶色谱渗透仪(GPC,日本岛津制作所)测定,两根GMH-HR-H色谱柱(东曹公司)串联,相对分子质量范围1×104~2×106,K值为0.017 7 mL/g,α值为0.735,温度25 ℃,四氢呋喃作为流动相,流速1.0 mL/min,标样为聚苯乙烯。
TPR微观结构采用红外光谱仪及核磁共振仪测定。使用美国BIO/IAD公司的FPS3000型红外光谱仪进行红外光谱测定,将聚环戊烯溶液涂在溴化钾盐片上,使溶液均匀分布于光束区,在室温下蒸发溶剂,使试样的透射率保持在30%~50%,扫描范围400~4 000 cm-1,扫描次数32次,分辨率2 cm-1。采用瑞士Bruker公司的DRX400MHz型核磁共振仪测定TPR的核磁共振谱图,溶剂CDCl3,25 ℃,用TMS定标。
TPR玻璃化转变温度Tg及熔融温度Tm采用美国TA公司MDSC2910型差示扫描量热仪测量,升温速率10 ℃/min,温度范围-150~80 ℃。
TPR凝胶含量测定方法为将放有一定量橡胶试样的不锈钢网小筐置于甲苯中,溶解24 h后取出放入120 ℃烘箱中烘干并称重,计算凝胶含量,不锈钢网筛孔直径为0.125 mm,凝胶含量按式(1)计算。
G=[(m2-m1) /m]×100%
(1)
式中,G为凝胶质量分数,m为试样质量,m1为小筐质量,m2为小筐与凝胶总质量。
2 结果与讨论
2.1 催化剂陈化与非陈化对环戊烯聚合反应的影响
环戊烯聚合早期采用二元催化体系,因受空气中水和氧的影响较大,聚合重复性差,之后采用添加活化剂的方法稳定催化剂活性[3,17]。本文采用酚类物质作为活化剂,催化剂的制备方法包括:1)催化剂不陈化,即主催化剂、活化剂与助催化剂溶液分别单独加入到聚合体系中;2)催化剂陈化,即将主催化剂与活性剂溶液在一定温度、一定时间内按比例混合后静置一定时间,然后将陈化后的催化剂与助催化剂分别加入聚合体系中。其他条件相同时催化剂制备方式对催化剂活性及聚合反应的影响见表1。
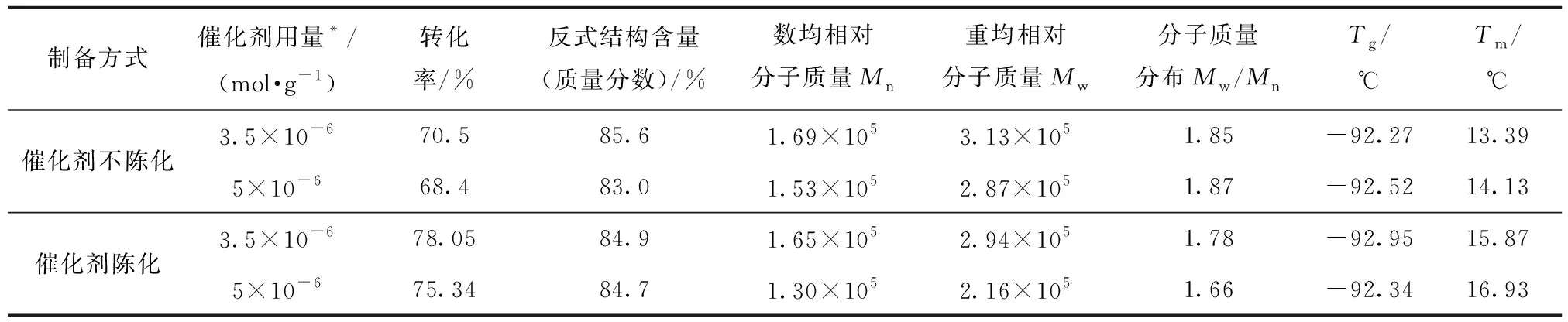
表1 催化剂陈化与非陈化对环戊烯聚合反应的影响Table 1 The effect of aging and non-aging of the catalyst on polymerization
由表1可以看出,使用钨盐作为环戊烯聚合催化剂时,不论催化剂陈化与否均可使环戊烯有效聚合,但催化剂陈化后活性更高,单体转化率也更高,最高可达78%以上。另外还发现,陈化后的催化剂室温下放置2个月活性基本维持不变,而未陈化的主催化剂溶液放置3 d左右即有少量沉淀出现,表明陈化后的催化剂具有非常好的稳定性。催化剂陈化后稳定性提高的原因是陈化产物已经不是原来两组分的简单混合,而是伴随着化学反应的发生,含羟基的活化剂与钨盐反应后生成了钨的烷氧基化合物,该化合物的形成不但降低了水、氧等有害杂质对活性中心的破坏,而且使活性中心在有机溶液中的溶解度大大提高,这样就使得催化活性中心数量增加,从而提高了催化剂的活性及稳定性[3,18]。催化剂陈化与否对聚环戊烯的反式结构含量、相对分子质量及其分子质量分布、玻璃化转变温度及熔融温度等的影响不大。此外,用该催化剂合成的聚环戊烯的反式结构含量、相对分子质量及其分子质量分布均较适宜,其Tg较天然橡胶(-72 ℃)更低,说明其有更好的低温性能,Tm与NR(约30 ℃)接近,与文献报道的聚环戊烯橡胶结构相当[18]。
2.2 催化剂用量对环戊烯聚合反应的影响
催化剂用量是评价TPR的生产成本及技术经济性的一个重要因素,实验考察了相同条件下催化剂用量对聚合反应的影响。由于在本文实验条件下TPR的反式结构含量、玻璃化转变温度及相对分子质量分布等受其他因素影响的变化不大,故暂不作考察,着重分析了催化剂用量对单体转化率及数均相对分子质量的影响,结果见图1。
由图1可看出,单体转化率不因催化剂用量的增加而升高,甚至出现降低,这与双烯烃聚合不同,反映出开环聚合的特点。当然催化剂用量不能太少,用量太少活性中心数量不够,但用量太大对提高催化剂活性及聚合转化率并无太大帮助,而且会造成催化剂浪费,这可能是因为催化剂用量太大会造成活性中心聚集从而使部分催化剂失活,与文献[3]结论一致。当催化剂用量减小至3.5×10-6mol/g之后,单体转化率基本保持稳定,因此通常采用催化剂用量为(2.5~3.5)×10-6mol/g。当催化剂用量为2.5×10-6mol/g时,催化剂活性以WCl6计为每mol钨可聚合4×105g环戊烯,此时的活性已经达到文献最佳值[3]。从图1还可以看出,随着催化剂用量的增加,数均相对分子质量先升高后降低,与聚合反应转化率的趋势接近,这可能是由于催化剂用量增大,活性中心数量增加,导致相应的数均相对分子量降低,与文献[3]结论一致。
2.3 聚合溶剂对环戊烯聚合反应的影响
聚合溶剂对聚合物溶液黏度、反应过程传质、聚合转化率、聚合物结构以及溶剂循环利用等均会造成不同程度的影响。实验考察了在其他条件相同时不同聚合溶剂对环戊烯聚合反应的影响,结果见表2。
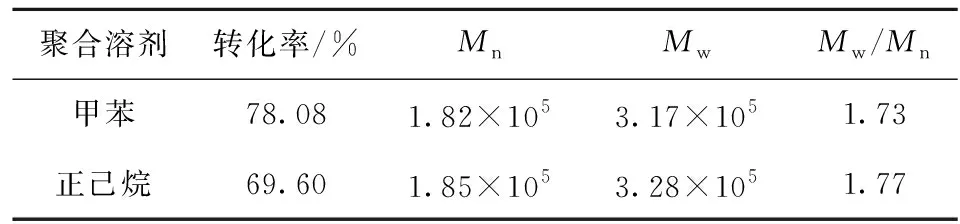
表2 聚合溶剂对环戊烯聚合反应的影响Table 2 The effect of polymerization solvent on cyclopentene polymerization
由表2可以看出,用甲苯作聚合溶剂的单体转化率高于正己烷,但正己烷无毒且沸点较低,有利于溶剂循环使用时的节能降耗,综合考虑采用正己烷作为环戊烯聚合反应的溶剂更合适。从表2还可看出,采用甲苯、正己烷作聚合溶剂制得的聚环戊烯数均相对分子质量在20万左右,分子质量分布在1.7~1.8之间,满足作为通用胶的要求[7]。
2.4 分子量调节剂与W的物质的量比对环戊烯聚合反应的影响
采用α-烯烃作为分子量调节剂,相同或相近条件下,分子量调节剂与W物质的量比对单体转化率及数均相对分子质量的影响见图2。
由图2结果可知,当不添加分子量调节剂时,聚合物的数均相对分子质量达60万以上,重均相对分子质量可达110万以上,聚合物中凝胶含量超过43%,此时聚合物溶液黏度太大导致其很难流动,工艺上很难实施。分子量调节剂对聚合物分子量的调节作用非常明显,当分子量调节剂与W物质的量比为2~4时,聚合物数均相对分子质量为(10~30)万,此时相对分子质量分布在1.7~2.3之间,凝胶含量小于1.0%,可满足通用橡胶对相对分子质量及凝胶含量的要求。由图2还可以看出,分子量调节剂与W物质的量比对聚合转化率有一定影响,但程度不大且没有一定规律。通常取分子量调节剂与W物质的量比为2~3。
2.5 Al与W的物质的量比对环戊烯聚合反应的影响
烷基铝化合物(简称Al)是环戊烯聚合催化剂的助催化剂,起到还原剂的作用。Al与W物质的量比决定着W盐的还原程度以及活性中心的数量,在相同条件下通过实验考察了Al与W物质的量比对聚合反应的影响,结果见图3。
由图3可以看出,当Al与W物质的量比为1~3时,单体转化率基本不变,随着Al与W物质的量比继续增大,单体转化率逐渐降低。随着Al与W物质的量比增大,聚合物分子量先增大然后逐渐减小,但与分子量调节剂相比,Al与W物质的量比的影响程度很小。较合适的Al与W物质的量比为1~3。这可能是因为合适的Al与W物质的量比会使主催化剂适度还原,有利于提高催化剂的催化活性,而Al与W物质的量比太大或太小,会导致主催化剂还原过度或还原不够,都会对单体转化率产生一定影响。至于Al与W物质的量比对聚合物分子量大小的影响目前还没有很好的理由予以解释。
2.6 聚环戊烯的结构
2.6.1微观结构
环戊烯在钨或钼催化剂作用下通过开环聚合得到聚环戊烯,聚环戊烯有顺式及反式链节结构,其顺反结构含量由催化剂种类及聚合工艺条件等决定,聚环戊烯的化学结构式见图4。环戊烯聚合反应机理由Dall’Asta等得到证明,开环点在环戊烯的双键处,得到的聚环烯烃约含85%左右的高分子量无环大分子及15%左右分子量为400~500的大环齐聚物,聚环戊烯的分子量可通过添加少量α-烯烃予以调整[17-19]。
聚环戊烯中双键的类型及数量常用红外光谱和核磁共振光谱测定,红外光谱法计算反式链节含量时由于所用特征频率和消光系数不同,计算公式也不同,导致所得反式结构含量有差异,其计算公式如式(2)、(3)[3,18],据文献[3]报道式(2)计算的结果低于式(3)。
(2)
(3)
式中,T为聚环戊烯的反式结构含量,D965为红外谱图中聚环戊烯的反式结构在965 cm-1处的吸收峰强度,D1 404、D720分别为聚环戊烯的顺式结构在1 404、720 cm-1处的吸收峰强度。
聚环戊烯的13C NMR共振谱图见图5,其中trans-α代表聚环戊烯中α位的反式结构碳原子,cis-α代表α位的顺式结构碳原子,β代表β位的碳原子。
本文在文献[6]基础上采用核磁共振波谱法对聚环戊烯橡胶进行了一维1H NMR和13C NMR碳谱分析,结合二维核磁法,对聚环戊烯橡胶顺反异构体的核磁谱峰进行归属,顺、反异构体比例Xtrans、Xcis计算公式为
Xtrans=Atrans/(Atrans+Acis)×100%
(4)
Xcis=100%-Xtrans
(5)
式中,Atrans为化学位移在32.3~33.5范围内的峰面积;Acis为化学位移在27.4~27.9范围内的峰面积。
根据式(4)、(5)计算得到聚环戊烯橡胶反式异构体含量,并与红外法结果进行了比对,结果见表3。

表3 聚环戊烯橡胶反式含量测试方法比较Table 3 Comparison of methods for determining the trans content of polycyclopentene rubber
由表3可看出,红外光谱法与核磁共振法相比结果偏高且相对误差较大,但二者的平行性较好。这是因为红外光谱法仅适用于组分简单、特征吸收带不重叠、浓度与吸收度呈线性关系的样品,且易在制样过程中引入误差,但此方法操作简单,可用于聚环戊烯橡胶结构的初步分析。而核磁共振碳谱分辨率高,对聚环戊烯橡胶顺、反异构体比例的测定影响因素少,可靠性高,适合于对聚环戊烯橡胶顺反异构体含量的定量分析。
2.6.2分子量及分子量分布
聚合物的相对分子质量及分子量分布不但对其应用性能有影响,而且还会影响其门尼黏度及加工性能。在环戊烯聚合过程中通过添加分子量调节剂可以实现聚合物分子量的调节,但分子量分布的变化不大。图6是典型的反式聚环戊烯橡胶的GPC谱图,图中TPR的峰值相对分子质量为28.3万,数均相对分子质量为17.8万,相对分子质量分布为1.75,相对分子质量及分布均较适宜。
2.6.3DSC谱图
TPR的DSC谱图如图7所示。由图7可看出,TPR的Tg小于-92 ℃,说明其在低温下仍有较好的弹性,其低温性能甚至好于NR;Tm在15 ℃左右,与NR的Tm接近,并且在-50~20 ℃间有一个较大的吸热峰,初步判断含有部分结晶,结晶含量仍有待进一步研究。由文献[18]知,TPR在室温下(20 ℃左右)不结晶,拉伸时会很快结晶,但可以通过调节顺反结构含量或添加增塑剂使其结晶速度与通用橡胶相近,满足通用橡胶基本性能的要求。
3 结论
(1)确定了用于TPR合成的催化剂制备工艺条件,即主催化剂溶液与活化剂溶液按比例混合并陈化,此时催化剂具有较高的活性及稳定性。甲苯及正己烷作为聚合溶剂均可使环戊烯有效聚合。
(2)当催化剂WCl6用量为(2.5~3.5)×10-6mol/g,分子量调节剂与W物质的量比为2~3,Al与W物质的量比为1~3时,单体转化率大于78%,TPR反式结构含量(质量分数)约为85%,玻璃化温度小于-92 ℃,数均相对分子质量在10万~30万之间,相对分子质量分布在1.5~2.5之间,满足通用橡胶对结构及分子量的要求。
猜你喜欢
中国生殖健康(2020年5期)2021-01-18 03:00:06
天然产物研究与开发(2018年10期)2018-11-06 07:43:52
天然产物研究与开发(2018年10期)2018-11-06 07:43:42
中国生殖健康(2018年5期)2018-11-06 07:15:56
食品与机械(2018年5期)2018-07-16 01:34:00
现代园艺(2018年3期)2018-02-10 05:18:22
中国民族医药杂志(2016年5期)2016-05-09 07:43:50
中国继续医学教育(2015年5期)2016-01-07 07:38:27
中国医学科学院学报(2015年5期)2015-03-01 04:03:35
天然产物研究与开发(2014年6期)2014-04-27 14:16:00
