
Keggin型钨硅酸-镍-联咪唑的合成及电催化性能
2020-10-09 12:09:40张澜萃李孟璇王秋雪
辽宁师范大学学报(自然科学版) 2020年3期
张澜萃, 李孟璇, 王秋雪
(辽宁师范大学 化学化工学院,辽宁 大连 116029)
多金属氧酸盐(polyoxometalates,POMs)是一类特殊的富氧金属簇合物,也称多酸,其结构和组成多变,具有优异的物理化学性质.POMs可被有机基团或金属配合物修饰,而通过合成设计可对金属离子、有机配体和POM进行选择,以构建不同结构、不同性能的无机-有机杂化材料.H2biim作为具有多个配位点及丰富配位形式的含氮有机分子,是配位化学、超分子化学及其相关领域的重要研究对象之一,因此将H2biim引入POMs中,可以形成许多新颖的结构,且能改变POM表面的一些性质[1-5].Keggin型POMs是多酸中研究最为广泛的一类,其阴离子基本单元是由12个MO6八面体围绕1个中心XO4四面体构成的饱和结构.因具有良好的稳定性和易修饰性,在催化、药物及材料科学等领域具有潜在的应用前景[6-10].近年来,以1∶12 Keggin型饱和阴离子为基本单元,通过与金属-有机配体配位形成支撑型或缠绕型结构的POMs不断被报道[11-15].该类化合物具有良好的催化性能,如龙腊生[16]等在2009年报道的Cu与有机配体4,4′-bpy共修饰的[SiW12O40]4-化合物用于催化乙苯氧化;2019年李健生[17]等使用Ag和H2biim共修饰的PW12作为催化剂进行电催化水氧化研究.因此,将H2biim引入Keggin型POM体系中以获得结构新颖、性能优异的无机-有机杂化材料具有研究前景.但此类合成反应中饱和POM和H2biim对pH值的变化敏感,副产物多,易生成沉淀,且H2biim对与金属配位的环境要求较高,因此探讨一条方便有效的合成路径是一项有意义的工作.采用常规水溶液和水热合成方法,利用自组装原理,以缺位Keggin结构多酸为起始物,尝试将过渡金属镍Ni(Ⅱ)和H2biim引入饱和Keggin型钨硅酸盐结构中,以构筑Ni(Ⅱ)和H2biim共修饰的基于Keggin结构POM的无机-有机杂化化合物,获得其单晶结构,并利用循环伏安(CV)法分别研究该类化合物在溶液中对H2O2和NaNO2的电催化还原性能.
1 实验部分
1.1 实验试剂和测试手段
参照文献[18]分别合成和表征三缺位Keggin结构钨硅酸钠Na10[SiW9O34]·18H2O和单缺位Keggin结构钨硅酸钠Na8[SiW11O39]·14H2O.所用原料为购买的分析纯试剂,未经处理直接使用.采用Vario Elcube元素分析仪;Bruker Smart APEX Ⅱ X射线单晶衍射仪(Mo Kα衍射源,λ=0.071 073 nm);Bruker AXS TENSOR-27 FT-IR傅立叶变换红外光谱仪(4 000~400 cm-1,KBr压片);Bruker AXS D8 X射线粉末衍射仪(Cu Kα衍射源,λ=0.154 18 nm);Pyris Diamond TG/DTA型热重-差热综合分析仪(空气气氛,10 ℃·min-1);CHI604B电化学工作站,三电极体系:玻碳电极为工作电极,Ag/AgCl为参比电极,铂丝为对电极.
1.2 化合物1的合成
称取0.30 g(0.11 mmol)Na10[SiW9O34]·18H2O溶于10 mL水中,依次加入2 mL 0.2 mol·L-1Ni(NO3)2溶液和0.03 g(0.23 mmol)H2biim,用稀HCl调节pH至3~4,室温下搅拌2 h,将反应液转移至内衬聚四氟乙烯的体积为30 mL不锈钢反应釜中,160 ℃恒温3 d后,冷却至室温,过滤,所得滤液在室温下缓慢蒸发,约6 h后得到深红色块状晶体,分离出晶体,待测.元素分析C24H42N16Ni2O49SiW12(化合物1),wt%(质量百分数):C 7.90,H 1.23,N 6.15(实测值);C 7.81,H 1.15,N 6.07(计算值).
1.3 化合物2的合成
将0.54 g(0.17 mmol)Na8[SiW11O39]·14H2O溶于20 mL 0.2 mol·L-1NaAc-HAc(pH=4.0) 缓冲溶液中,依次加入1 mL 0.2 mol·L-1Ni(NO3)2溶液和0.03 g(0.23 mmol)H2biim,室温下搅拌1 h后过滤,所得滤液在室温下缓慢蒸发,3 d后得到红色片状晶体,分离出晶体,待测.元素分析C24H56N16Ni2O56SiW12(化合物2),wt%(质量百分数):C 7.63,H 1.55,N 5.95(实测值);C 7.55,H 1.48,N 5.87 (计算值).
1.4 化合物1和化合物2的晶体结构测定
选取大小适合、形状规则的化合物1和化合物2晶体样品,分别在2.055≤θ≤24.996和2.014≤θ≤24.997范围内收集衍射数据,衍射数据经LP因子和原子吸收校正,用直接法得到所有非氢原子坐标并经最小二乘法修正,水分子上的所有氢原子直接加在最后分子式中,其他氢原子坐标采用几何加氢的方法得到,所有计算采用SHELXTL-2014程序完成[19].化合物1和化合物2的晶体学数据列于表1中.

表1 化合物1和化合物2的晶体学数据表

表1(续)
1.5 化合物1/2电催化还原H2O2
以电催化还原H2O2为模型反应,探讨了化合物1/2作为电催化剂的催化性能.具体操作为:将化合物1/2溶于1 mol·L-1H2SO4中,浓度为1×10-4mol·L-1,分别依次加入0、1、2 mmol·L-1H2O2,在扫速为50 mV·s-1时,利用CHI604B电化学工作站,在玻碳电极为工作电极,Ag/AgCl为参比电极,铂丝为对电极的三电极体系中进行循环伏安(CV)测试.
1.6 化合物1/2电催化还原NaNO2
采用与1.5相同的三电极体系和相似测试方法,同时考察了2例化合物电催化还原NaNO2的性能.即以1 mol·L-1H2SO4为电解质溶液,化合物1/2为电催化剂,其浓度均为1×10-4mol·L-1,当NaNO2的浓度为0、1、2 mmol·L-1,扫速为50 mV·s-1时,分别测试化合物1/2电催化还原不同浓度NaNO2的CV曲线,依据电流的变化和大小评价其催化性能。
2 实验结果与讨论
2.1 合成讨论
在2例化合物的合成过程中,采用不同的反应前驱体,通过其他条件控制,包括反应溶剂、pH和晶化等条件,可以得到主体POM阴离子结构相同的化合物.如化合物1是以三缺位Keggin结构的Na10[SiW9O34]·18H2O为前驱体,用稀HCl调节溶液pH为3~4,在水热条件下合成、常温下较快晶化得到的晶体产物.另外,当采用饱和Keggin结构钨硅酸和Ni(Ⅱ)-H2biim配合物为起始原料时,利用AB溶液合成法也可得到化合物1[20],但得到的晶体化合物量较少,主要是粉末形式,溶解度较小.如果采用单缺位的Keggin结构Na8[SiW11O39]·14H2O为POM前驱体时,即使在pH=4.0的NaAc-HAc缓冲溶液中,单缺位结构也不能保持,也将转化为饱和Keggin结构,在常规条件下合成、常温下缓慢晶化得到了晶体化合物2.2例化合物的合成说明,除了pH是影响缺位结构稳定性的重要因素外,晶化条件包括晶化介质、晶化时间等也是重要影响因素.
2.2 晶体结构描述


图1 化合物1(a)和化合物2 (b)的多面体-球棍图
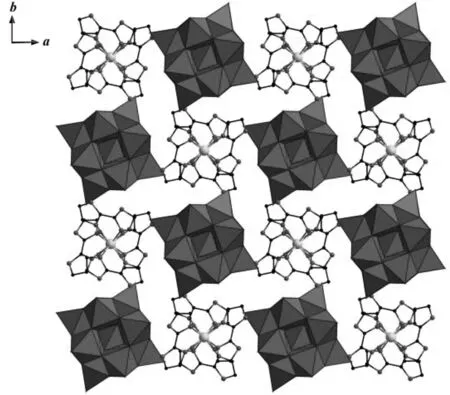
图2 化合物1沿c轴的堆积图(八面体:WO6;四面体:SiO4;大球:Ni;中球:N;小球:C、O.删除所有H)

图3 化合物2沿c轴的堆积图(八面体:WO6;四面体:SiO4;大球:Ni;中球:N;小球:C、O.删除所有H)
2.3 化合物的表征
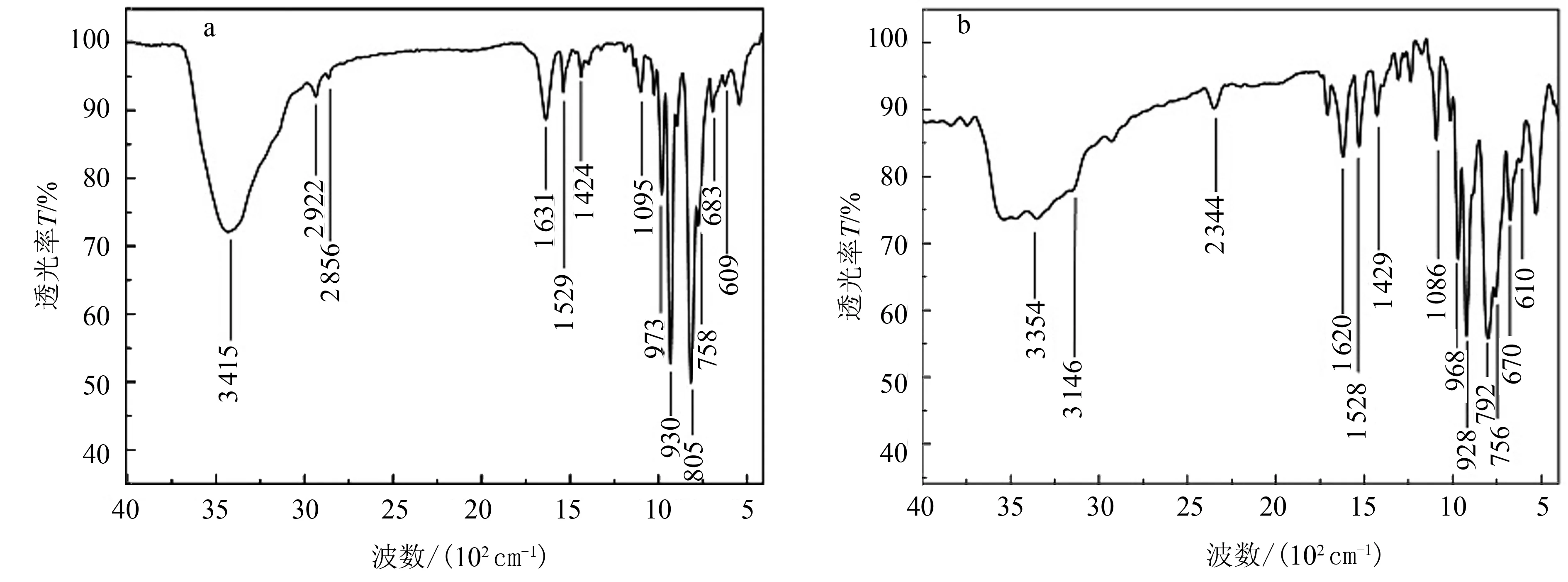
图4 化合物1(a)和化合物2(b)的IR光谱
2.3.2 粉末X射线衍射(PXRD)分析 图5a和5b分别为化合物1和化合物2的PXRD与SCXRD拟合对比图.2个图中的PXRD实测值和SCXRD拟合值的峰位均吻合良好,说明化合物1和化合物2基本为纯相,峰强度的不同可能是由于粉末样品测定取向不同所致.

图5 化合物1(a)和化合物2(b)的PXRD衍射图
2.3.3 热重(TG)分析 化合物1和化合物2在室温至1 000 ℃的TG-DTA曲线如图6所示.从图6a中可以看出,化合物1在室温至667 ℃之间经历3步连续失重过程,总失重18.95%,与理论失重(18.93%)基本吻合,对应于失去所有的结晶H2O、配位H2O和H2biim分子.在667 ℃后出现平台,至1 000 ℃再无失重发生,说明多酸阴离子骨架依然保持.图6b中的TG曲线显示4步连续失重,约850 ℃以后出现平台,从室温开始至此温度化合物2的实际总失重为33.45%.而理论总失重(21.86%)包括失去6个结晶H2O分子、4个配位H2O分子、2个CH3COOH分子和4个H2biim分子,明显低于实际失重,说明化合物2的多酸阴离子骨架已坍塌.与化合物1相比,化合物2在约700 ℃后仍进一步失重,因此,化合物1具有较高的热稳定性.

图6 化合物1(a)和化合物2(b)的TG-DTA曲线
2.3.4 循环伏安(CV)分析 图7a给出了化合物1在1 mol·L-1H2SO4溶液中不同扫速下的CV图.从图7a可以看出,在900~-200 mV,扫描速度10~50 mV·s-1范围内,峰电流均增加,峰电位基本不变,说明化合物1在1 mol·L-1H2SO4溶液中比较稳定.另外,CV曲线显示了3对明显的氧化-还原峰(还原电势Epc:Ⅰ,-577.50 mV;Ⅱ,-706.11 mV;Ⅲ,-787.37 mV;氧化电势Epa:Ⅰ′,-543.49 mV;Ⅱ′,-672.09 mV;Ⅲ′,-753.35 mV),中点电位Emid=(Epc+Epa)/2分别为-560.50、-689.27、-770.36 mV,峰电位差(ΔE)分别34.01、34.03、34.02 mV,氧化-还原峰Ⅰ-Ⅰ′、Ⅱ-Ⅱ′和Ⅲ-Ⅲ′分别对应化合物1的2、2、2电子的氧化-还原过程.
图7b给出了化合物2在1 mol · L-1H2SO4溶液中不同扫速下的可重复的CV图.从图7b可以看出,在800~-800 mV,扫描速度50~250 mV·s-1范围内,峰电流均增加,峰电位基本不变,说明化合物2在1 mol · L-1H2SO4溶液中是比较稳定的.CV曲线显示了3对明显的氧化-还原峰(Epc:Ⅰ,332.15 mV;Ⅱ,-585.62 mV;Ⅲ,-757.39 mV;Epa:Ⅰ′,390.66 mV;Ⅱ,-523.77 mV;Ⅲ′,-699.09 mV),Emid分别为361.41、-580.4、-728.24 mV,ΔE分别为58.51、61.85、58.3 mV,氧化-还原峰Ⅰ-Ⅰ′、Ⅱ-Ⅱ′和Ⅲ-Ⅲ′分别对应化合物2的1、1、1电子的氧化-还原过程.
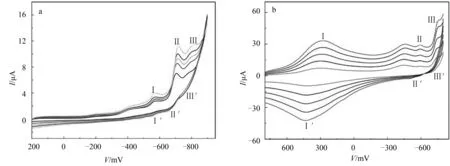
图7 化合物1(a)和化合物2(b)在1 mol· L-1 H2SO4溶液中不同扫速下的CV曲线
2.4 化合物1和化合物2的电催化性能
POMs是一种很有前途的电催化剂,可以在不改变初始结构的情况下经历一系列连续和可逆的多电子氧化-还原过程.图8、图9为化合物1和化合物2在1 mol · L-1H2SO4中扫速为50 mV·s-1时的电催化还原H2O2和NaNO2的CV曲线.
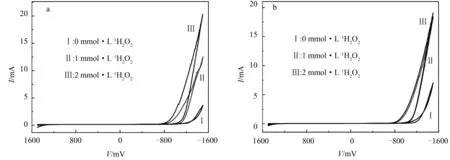
图8 化合物1(a)和化合物2(b)在1 mol·L-1 H2SO4溶液中对不同浓度H2O2的电催化还原曲线
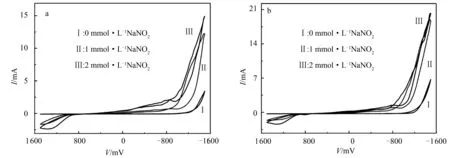
图9 化合物1(a)和化合物2(b)在1 mol·L-1 H2SO4溶液中对不同浓度NaNO2的电催化还原曲线
由图8、图9可以看出,当电解质溶液中仅含POM催化剂,即不含H2O2或NaNO2(0 mol·L-1H2O2或NaNO2)时,化合物1和化合物2的催化电流分别为3.64、7.08 mA;当加入1 mmol·L-1H2O2时,显示催化电流分别为12.35、18.33 mA;在加入2 mmol·L-1H2O2后催化电流分别为20.22和19.09 mA.另外,图9a和图9b显示化合物1和化合物2在催化还原1 mmol·L-1NaNO2时,其催化电流分别为12.37、19.11 mA,当浓度增大到2 mmol·L-1NaNO2后催化电流分别为15.10、20.51 mA.
实验结果表明,随着不同浓度H2O2或NaNO2的加入,氧化-还原电对的峰电流增大,还原峰电流的增加尤为突出,说明化合物1和化合物2对H2O2和NaNO2具有显著的电催化作用.
根据电催化效率(CAT)公式可计算出2例化合物电催化还原H2O2和NaNO2的CAT值[24]:
CAT=100% × [Ip(POM,底物)-Ip(POM)]/Ip(POM).
(1)
其中,IP(POM,底物)和IP(POM)分别是化合物在有催化底物和没有催化底物存在情况下的峰值电流.根据图8、图9中CV峰电流值,通过式(1)计算得到化合物1和化合物2电催化还原H2O2的CAT分别为455.49%和169.63%,电催化还原NaNO2的CAT分别为314.84%和189.69%.计算结果表明,化合物1具有更高的催化效率.
3 结 论
采用常规水溶液和水热合成的方法,通过常温进一步晶化,获得2例基于饱和Keggin型POM的无机-有机杂化晶体化合物.2例化合物的阴离子结构基本相同,但配阳离子中与Ni相连的2个H2biim形成的平面夹角不同.另外,所含结晶H2O分子数不同,化合物2中还含有2个游离的CH3COOH分子,其热稳定性低于化合物1.化合物1具有更高的电催化还原H2O2和NaNO2的催化效率,这可能是由于2例化合物分子的空间堆积形式和H2biim平面间形成的夹角差异所致,该电催化还原反应的催化机理有待进一步探讨.
猜你喜欢
辽宁化工(2022年8期)2022-08-27 06:02:54
布达拉(2022年8期)2022-05-30 10:48:04
建材发展导向(2021年13期)2021-07-28 07:14:40
民主与法制(2020年16期)2020-08-24 06:54:46
陶瓷学报(2019年6期)2019-10-27 01:18:38
传媒评论(2018年8期)2018-11-10 05:22:12
中国有色金属学报(2018年2期)2018-03-26 07:58:37
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
中国资源综合利用(2016年7期)2016-02-03 03:00:13
无机化学学报(2014年3期)2014-02-28 17:30:48
