
云南金花茶转录组序列分析及功能注释
2020-09-30 02:51培尧
经济林研究 2020年3期
, , , ,b,,培尧
(1.西南林业大学 a.西南山地森林资源保育与利用教育部重点实验室;b.国家林业和草原局西南地区生物多样性保育重点实验室,云南 昆明 650224;2.毕节市林业科学研究所,贵州 毕节 551700;3.云南省大围山国家级自然保护区河口管理分局,云南 河口 661399)
云南金花茶Camellia fascicularisH.T.Chang为山茶科Theaceae 山茶属Camellia金花茶组的常绿木本植物[1]。其现有野生资源仅600 株左右,属于我国二级保护植物,同时也被云南省认定为特有极小种群植物[2]。云南金花茶间断分布于河口及其周边个旧市和马关县的沟谷或山坡海拔400 ~1 000 m 湿润肥沃的土壤中,耐贫瘠[3]。云南金花茶花形娇艳多姿,呈金黄色,除具有园林观赏价值外,还具有降血脂、抗氧化、抑制癌细胞等功效[4-6]。近年来,有关云南金花茶的研究报道主要集中在繁育技术、离体培养体系建立、保健功效、营养成分分析以及化学元素研究[7-11]等方面,而在分子水平的研究少有报道,仅见本课题组利用SSR 分子标记技术对云南金花茶遗传多样性的分析[12]。
随着分子生物学相关知识和技术的快速升级,利用转录组测序(RNA-seq)技术获得基因信息,对植物进行生物信息学分析的方法得到广泛应用。使用RNA-seq 技术可在获得大量物种注释序列信息的同时,挖掘出生物重要的功能基因,因此该技术成为研究缺乏功能组学和基因组学信息的物种优良性状和基因功能的重要手段[13-15]。伴随高通量测序技术的不断进步与完善,RNA-seq 成本不断降低,核苷酸的检测也更精准和快捷[16-19]。目前,该技术在前基因组学和后基因组学的研究中均有应用。魏开发等[20]对火龙果Hylocereus undatus进行RNA-seq,研究不同发育阶段中花芽、果实和枝条基因的表达情况,并将组装得到的Unigene 进行注释,结果发现,在火龙果不同组织中分别有多个特异表达的Unigenes。宋猜等[21]对采于花芽萌动前后3 个发育时期的仁用杏Armeniaca vulgaris花芽进行转录组测序,试验数据可提供大量仁用杏开花相关基因的信息,可为研究仁用杏成花分子机制及解决仁用杏花期冻害问题提供理论依据。张贤等[22]通过RNA-seq 对芒草Miscanthus sinensis的74 134 条Unigenes 进 行不同功能领域数据库注释,其结果可为进一步鉴定中国芒草的功能基因提供参考。另有学者对百香果Passiflora edulis[23]、云锦杜鹃Rhododendron fortunei[24]、桐花树Aegiceras corniculatum[25]进行了相关研究。目前,仅见叶鹏等[12]对云南金花茶转录组微卫星序列的分布与特征进行了探讨,而有关其基因功能注释、代谢途径和代谢通路方面的分析未见报道。本研究中对云南金花茶进行高通量转录组测序,获得大量原始数据的同时,对其进行拼接与组装处理,建立RNA-seq 数据库,并将处理后的数据在7 大公共数据库进行功能注释、代谢途径和相关通路分析,以期为云南金花茶乃至山茶属植物功能基因的探索提供一定的理论参考。
1 材料与方法
1.1 试验材料
在云南省河口县(103°97′E,22°52′N)海拔1 036 m 的阳坡地带采集云南金花茶植株,引种到西南林业大学温室大棚中。选用1 份云南金花茶幼嫩叶片作为试验材料,用锡箔纸包好放入液氮中,备用。
1.2 试验方法
1.2.1 转录组测序
首先提取云南金花茶RNA,然后构建cDNA数据库,继而利用Illumina HiseqTM 2000 平台对云南金花茶进行转录组测序。此部分工作由北京诺禾源科技股份有限公司完成。
1.2.2 序列组装
RNA-seq 测序完成后,统计raw reads 的数量和长度。原始数据中含有低质量序列、重复冗余序列、接头和无法确定碱基信息的序列,必须将上述序列去除,以获得clean reads,继而统计clean reads 的数量、总长度、处理后不确定序列所占比例、GC 碱基所占比例、N50(拼接转录本不小于总长50%的长度)以及Q20(处理后质量高于20的碱基)所占比例等。对clean reads 通过Trinity Software(http:///trinityrnaseq.Github.io/) 进 行de novo 组装。首先利用clean reads 之间的overlap 将其向两边伸展形成序列重叠克隆群(contig),再依据序列双末端的信息对contig 进行再次连接,得到该样品的Transcript,去除Transcript 冗余reads 获得Unigene 后,进行Transcript 和Unigene的分布和长度分析[26]。
1.2.3 基因功能注释、分类及生物学通路分析
将处理得到的Unigene 在7 个不同功能领域的公共数据库中进行基因功能注释和分类分析,从而获得较全面的云南金花茶基因功能信息。数据库包括:Nr(Non-Redundant Protein Database,非冗余蛋白数据库)、Nt(Nucleotide Sequence Database,核酸序列数据库)、Pfam(Pfam Protein Sequence Database,Pfam 蛋白序列数据库)、KOG(euKaryotic Ortholog Groups,真核生物蛋白直系同源数据库)、Swiss-Prot(Swiss-ProtProtein Database,Swiss-Prot 蛋白质序列数据库)、KEGG(Kyoto Encyclopedia of Genes and Genomes,基因组百科全书)、GO(Gene Ontology,基因本体数据库)[27]。使用BLAST 软件将Unigene 在Nr、Nt、Swiss-Prot 等数据库中进行比对(e-value <1×10-5),获取相关基因注释。比对到Nr 数据库中[28],从而获取云南金花茶基因序列相似性和物种分布信息。依据Nr 中注释的结果,在Blast2GO 数据库比对,得到GO 功能注释信息[29]。GO 数据库包括3 大类别,分别为生物过程、分子功能与细胞组分,以此可以宏观解读云南金花茶基因功能的分布及特征[30]。将Unigene比对到KOG 数据库中,并按可能的功能对获得结果的Unigene 进行分类与统计;另外,对Unigene进行KEGG 数据库相关通路(包括细胞过程、遗传信息处理、新陈代谢、环境信息处理、有机系统5 大类别)分析,了解云南金花茶的代谢通路以及各通路之间的关系[31]。
1.2.4 云南金花茶转录组Unigene 的CDS 预测
将Unigene 序列依次比对到Nr(https://www.ncbi.nlm.nih.gov/)、Swiss-Prot(http://www.ebi.ac.uk/uniprot/)、KEGG(http://www.genome.jp/kegg/)、KOG(http://www.ncbi.nlm.nih.gov/COG/)等蛋白数据库中,对于未比对上或未预测到结果的序列,使用ESTScan(3.0.3)软件进行预测。
2 结果与分析
2.1 云南金花茶RNA-seq 及de novo 组装结果
通过RNA-seq,共得到云南金花茶57 051 836条原始序列。将原始序列中的接头(dadpter)、低质量reads、重复冗余以及不确定碱基含量超过10%的读序经处理后,获得54 817 600条有效序列,总长为8.22 Gb,Q20、Q30(处理后质量高于30的碱基)高质量序列分别占96.39%和91.28%,GC 含量占总碱基数的44.54%,碱基错误率为0.02%,说明由高通量测序平台获得了较高数量和质量的云南金花茶序列,有利于后续数据的组装,满足后期生物信息学的研究。得到的clean reads经de novo 组装后,共获得155 011 条Transcript,这些Transcript 经进一步组装之后,得到95 979 条Unigenes,序列信息达107 907 727 nt。对Transcript的序列长度分析结果表明,其平均长度是807 nt,N50是1 411 nt。其中,以200~500 nt的短序列居多,有85 904 条,占总数的55.42%;500 ~1 000 nt长度的序列为30 871 条,占总数的19.92%;1 000 ~2 000 nt 长度的序列为23 853 条,占总数的15.39%;大于等于2 000 nt 长度的序列占总数的9.28%(图1A)。Unigene 分析统计结果表明,其平均长度为1 124 nt,N50 为1 660 nt,其中1 000 ~2 000 nt 的序列占总序列的24.84%,超过2 000 nt 的序列占14.99%。通过对高通量RNA-seq得到的大量序列进行处理,经组装后Unigenes 数据的完整性明显提高,可进行下一步的分析统计(图1B)。
2.2 云南金花茶转录组Unigene 的功能注释及分类
将获得的95 979 条Unigenes 通过BLAST软件在7 大数据库进行比对, 共有63 888(66.56%)条Unigenes 获得注释。其中,在Nr(e-value ≤1×10-5)注释成功 的Unigenes 有58 830 条,占Unigenes 总数量的61.29%;在Nt(e-value ≤1×10-5)注释成功43 623 条,占总数的45.45%;在KEGG(e-value ≤1×10-10)注释成功的有23 214 条,占总数的24.18%;在Swiss-Prot(e-value ≤1×10-5)注释成功的有44 315 条,占总数的46.17%;在Pfam(e-value ≤0.01)注释成功的有41 096 条,占总数的42.81%;在GO(e-value ≤1×10-6)注释成功的有41 905 条,占总数的43.66%;在KOG(e-value ≤1×10-3)注释成功的有23 499 条,占总数的24.48%。在7 大数据库中均能得到成功注释的序列数目为11 933条,占总数的12.43%,其中63 888 条序列至少在1 个数据库中注释成功,占总数的66.56%。

图1 云南金花茶转录组组装序列长度分布Fig.1 Length distribution of assembly transcript andunigenes for C.fascicularis
2.2.1 云南金花茶转录组Unigene 的Nr 功能注释
通过Nr 库比对,云南金花茶有58 830 条Unigenes 在Nr 数据库中找到相似序列,注释匹配的物种主要有葡萄Vitis vinifera、中粒咖啡Coffea canephora、可可树Theobroma cacao、荷花Nelumbo nucifera、芝麻Sesamum indicum这5类,其中获得注释基因最多的是葡萄,有29.9%,中粒咖啡、可可树、荷花、芝麻分别仅占5.6%、5.3%、4.8%、4.7%,其余49.7%的注释基因分布于其他物种。从这些注释的信息中可以得出,云南金花茶的大部分序列均可以在被子植物中得到相应的匹配。从e-value 分布(图2A)可以看到,有44.2%的e-value 分布于l×10-100~l×10-45,有30%的e-value 分布于l×10-45~l×10-5,当e-value为0 时占25.8%。此外,有49.5%的序列相似度可达80%~95%,甚至有9.3%的序列相似度达到95%~100%,仅7.7%的序列相似度在60%以下,可以看出物种的序列相似度较高(图2B)。总体而言,从e-value 和序列相似度分布情况可看出,云南金花茶在Nr 数据库中比对的匹配度较高,但是由于缺乏云南金花茶一些基因组及转录组信息,导致部分Unigene 在数据库中未得到匹配。
2.2.2 云南金花茶转录组Unigene 的GO 功能注释
根据Nr 注释成功的基因进行GO 功能分类注释,其结果如图3所示。分析结果表明,共有41 905 条Unigene 注释了224 129 个GO功能,占Unigenes 总数量的43.66%。按3 大功能类别划分,生物过程功能类别基因序列为107 044 条,占总数的47.76%;细胞组分功能类别65 990 条,占总数的29.44%;分子功能类别51 095 条,占总数的22.80%。由此可知,在生物过程功能类别中所注释的基因比例最大。3 个功能大类进一步可划分为56 个GO 功能亚类,分别包括25、21 和10 个亚类。在生物过程包含的25 个功能亚类中,获得注释偏多的分别是代谢过程、细胞过程、单一有机体过程,分别占该类型的20.970%、22.570%和16.750%,细胞聚合过程所得到注释的比例最少,仅有0.008%。在细胞组分类别中,细胞和细胞部分所得到的注释居多,分别为12 867 和12 865 条,均约占细胞组分这一大类的19.500 0%,而细胞外基质组分、拟核、共质体得到注释较少,分别占0.006 1%、0.007 6%、0.006 1%。分子功能类别中,结合、催化活性得到的注释较多,各占所属分类总数的46.830 0%和38.100 0%,而金属伴侣蛋白活性所得注释最少,仅占0.005 9%。上述GO 功能注释结果显示了云南金花茶叶片中基因表达的基本情况,可以看出,在56 个功能亚类中,生物过程中代谢活动相关的基因量较多,说明云南金花茶有着较强的代谢能力。

图2 云南金花茶转录组Unigenes 的Nr 注释分类Fig.2 Annotation category on unigenes Nr in transcriptome of C.fascicularis
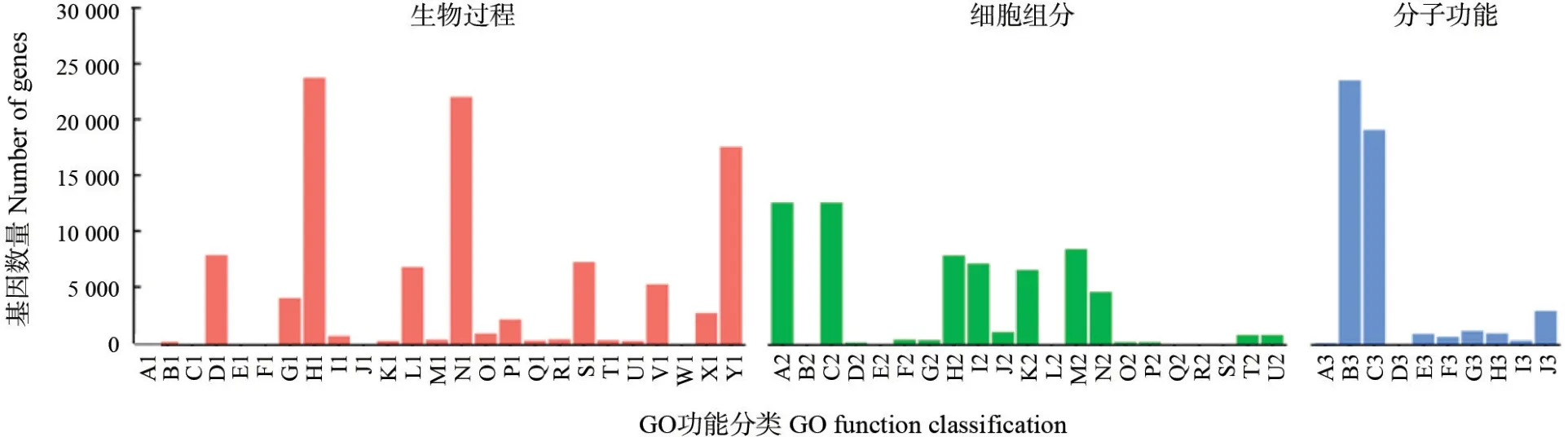
图3 云南金花茶转录组Unigene 的GO 功能分类Fig.3 GO classification of unigenes in transcriptome for C.fascicularis
2.2.3 云南金花茶转录组Unigene 的KOG 功能注释与分类
将获得的Unigene 进行KOG 蛋白数据库分类注释,其结果如图4所示。分析结果表明,在KOG 中有23 499 条Unigenes 能够匹配,占总数的24.480%,共获得KOG 功能注释信息26 430 个,根据比对结果可分为26 个功能大类,包括能量产生、转化,次生代谢物的生物合成、加工、运输等不同类别的基因表达。其中,主要为一般功能预测基因,占总数的16.620%;其次是翻译后修饰、蛋白质转化和分子伴侣基因,占总数的11.630%;信号传导机制(8.310%),转录(5.420%),RNA 加工和修饰(5.360%),胞内运输、分泌和膜泡运输(5.310%)的功能基因也占有较高的比例。而胞外结构所获得的功能注释信息最少,仅有54 个,占总数的0.020%。可见,云南金花茶在转录、翻译和蛋白质运输等方面的基因表达量较多。此外,还有1 个未知蛋白,不能获知其具体生物学功能,占总数的0.004%。
2.2.4 云南金花茶转录组Unigene 的KEGG 代谢通路分析
将获得的Unigene 比对到KEGG 数据库中,共有23 214 条Unigenes 获得注释,占总Unigenes数量的24.18%。根据其涉及的代谢通路可将云南金花茶Unigenes 归为5 大类别和19 个亚类,其结果如图5所示。

图4 云南金花茶转录组Uingenes 的KOG 功能注释分布Fig.4 KOG functional annotation distribution of unigenes in transcriptome for C.fascicularis
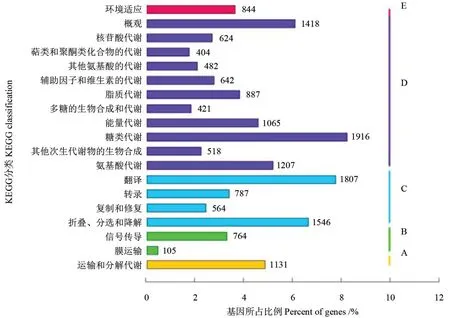
图5 云南金花茶转录组Unigene 的KEGG 分类Fig.5 KEGG classification of C.fascicularis Unigene
通过对图5中相关通路分类下Unigenes 的具体分析统计发现:5 大类别中代谢通路所占比例最多,有9 584 条Unigenes,占总数的55.94%;其次是遗传信息处理相关的通路,占27.46%;而环境信息处理和细胞过程中相关的通路,皆占6.60%;有机系统相关的通路最少,仅占4.93%。将5 大类别进一步细分为亚类,其中代谢相关的通路可分为11 个亚类,以糖类代谢居多,占注释Unigenes 总数的19.99%。其次为整体映射相关的通路,占总数的14.80%,萜类化合物和聚酮化合物的代谢相关通路最少,所占比例仅为4.22%。另外,遗传信息处理相关的通路分为4 个亚类,翻译相关的通路最多,占总数的38.41%;其次为折叠、分选和降解通路,占32.87%;复制和修复通路所占比例最少,仅为11.99%。在环境信息处理通路中,仅包括2 个亚类,以信号传导通路居多,占87.92%。细胞过程和有机系统相关的通路均仅1 个亚类,分别占6.60%和4.93%。在KEGG 代谢通路分析结果中,代谢通路类别所获得的注释基因最多,表明云南金花茶在这一时期有较强的代谢活动。
2.3 云南金花茶转录组Unigene 的CDS 预测
蛋白数据库的比对结果显示,有60 939 条Unigene 比对到蛋白库中,另预测到有26 428 条CDS,其长度分布如图6所示。从blast 比对得到的CDS 长度分布(图6A)可看出,达到1 000 nt以上长度的CDS 序列,占CDS 总数的30.59%,其中1 000 ~2 000 nt 长度的CDS 序列占总数的22.940%,2 000 ~9 000 nt 长度的CDS 序列占总数的7.630%,9 000 nt 以上长度的CDS 序列占0.260%。通过ESTScan 预测的CDS 长度集中分布于100 ~500 nt,占82.100%,有极少数序列的长度在9 000 nt 以上,占0.004%(图6B)。
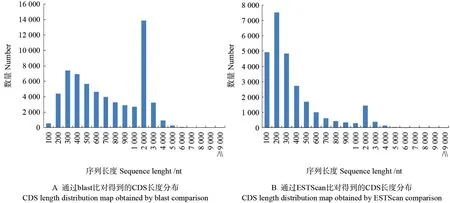
图6 云南金花茶转录组Unigene 的CDS 序列长度分布Fig.6 CDS length distribution of transcriptome for C.fascicularis
3 结论与讨论
采用Illumina HiSeq 2000 高通量测序可以同时完成前基因组学研究(测序和注释)以及后基因组学(基因表达及调控,基因功能,蛋白和核酸相互作用)研究。目前,此项技术已被应用于铁皮石斛Dendrobium officinale[32]、马尾松Pinus massoniana[33]、云南松Pinus yunnanensis[34]、红 豆杉Taxus chinensis[35]等多个物种基因组的分析。鉴于此,本研究中采用RNA-seq 技术对云南金花茶进行测序,获得95 979 条Unigene,平均长度为807 bp,结果与其他茶科植物如油茶Camellia oleifera[36]、紫芽茶树Camellia sinensis(Linn.) O.Kuntze[37]、“紫鹃”茶树Camellia sinensisvar.Zijuan[38]等相比,拼接完整性较好。从整体上看,通过对云南金花茶Unigene 总长、GC 含量、碱基正确率、序列Q20 的分析,测序获得的序列质量较高,数量较多,可为后续云南金花茶基因功能分析、分子标记开发、代谢通路等方面的研究提供参考。
通过Nr 数据库比对,有58 830(占总数的61.29%)条序列在不同物种均有相应的注释,其中注释匹配的物种主要为葡萄,有29.9%,其次为中粒咖啡、可可树等。从这些成功匹配的物种可以看出,云南金花茶的大部分序列均可以在被子植物中得到匹配。通过KOG 数据库比对,注释成功的KOG 功能信息为26 430 个,将其分为26 个功能大类,一般功能参与的基因最多,其次是翻译后修饰、蛋白质转化和伴侣基因。这与蒋会兵等[37]经研究得出的紫芽茶树转录组KOG 功能注释(26 个KOG 功能类别)分布大体一致。在KOG 注释中有0.004%的未知蛋白,难以确定其具体生物功能,可能是注释信息不完善所导致,这种情况在其他物种转录组分析中也有出现,如金钱松Pseudolarix amabilis[39]、云南松[34]、文冠果Xanthoceras sorbifolia[40]等。通过GO 数据库比对,共获得224 129 个GO 功能信息,按其具体的序列信息又可分为3 个大类和56 个亚类,其基因主要分布于细胞过程、代谢过程和单一有机体过程,且在56 个功能亚类中,生物过程中代谢活动相关的基因量较多,由此可看出,金花茶的代谢能力较强。根据KEGG 代谢通路分析结果,共有23 214(24.18%)条序列注释成功,按照注释结果可将其划分为5 大类别和19 个亚类,其中代谢通路相关基因所占比例最高,表明云南金花茶在整个时期均有较强的代谢活动。其基因注释分布特征与李明玺等[41]对靖安白茶转录组KEGG 注释结果基本一致。将云南金花茶Unigene 通过ESTScan进行预测,共获得26 428 条CDS,其长度集中分布于100 ~500 nt,而蔡年辉等[34]对云南松转录组Unigene 的CDS 预测结果显示其长度集中分布于200 ~1 000 nt,说明不同物种间存在较大差异。
通过7 个不同功能领域的基因蛋白数据库注释,可以看出云南金花茶所含基因信息丰富,通过分析所有注释信息,可以更深层次地探索其基因组信息和基因分布情况。尽管这些Unigenes 序列并未覆盖整个云南金花茶蛋白编码区,但所注释成功的基因仍有助于云南金花茶功能基因的挖掘和利用,以及为山茶属植物遗传育种等方面的研究提供理论参考。在注释中还有33.44%的Unigenes 未注释成功,这些Unigenes 序列可能为其他未编码RNA 序列和未含有蛋白质功能信息的序列,也可能是因为基因数据库中信息不足所导致。本研究结果也可为后续云南金花茶基因组水平的研究及遗传改良等方面的研究提供一定的参考,且可为云南金花茶的分子标记开发以及抗逆机理研究提供数据。
猜你喜欢
黄河之声(2021年18期)2021-12-14
中国生殖健康(2020年4期)2021-01-18
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
江苏农村经济(2019年5期)2019-01-14
读友·少年文学(清雅版)(2018年8期)2018-12-06
中国生殖健康(2018年4期)2018-11-06
数学大王·低年级(2017年10期)2017-10-31
边疆文学(文艺评论)(2017年5期)2017-06-07
军事历史(1985年2期)1985-01-18
