
介孔TiO2晶须-γ-Al2O3复合载体催化剂的制备及对二苯并噻吩的加氢脱硫性能
2020-09-27 09:11杨祝红花泽林李力成
石油学报(石油加工) 2020年4期
岳 凡,李 蒙,杨祝红,花泽林,李 龙,李力成
(1.南京工业大学 化工学院,江苏 南京 210009;2.南京林业大学 化学工程学院,江苏 南京 210037)
近年来,环境问题逐渐受到大众的广泛关注,中国出台的相关法规对污染排放控制也随之愈发严格,其中GB 19147—2016《车用柴油》标准要求燃料油中的硫质量分数低于10 μg/g,这对目前工业上广泛使用的油品加氢脱硫技术(HDS)提出了更高的要求,而催化剂性能的优劣是决定该技术的核心。
在众多催化剂调控技术中,开发新型载体材料是提升加氢脱硫催化剂性能较为简略的有效途径之一。目前最为广泛应用的加氢脱硫催化剂载体是γ-Al2O3,其具有高比表面积、高热稳定性和良好的孔结构等特点[1-2]。但是γ-Al2O3与活性组分间有强相互作用,容易形成尖晶石结构的稳定物质,这类物质不利于硫化形成催化活性中心[3],导致所制得的催化剂脱硫性能下降,因此,仅以γ-Al2O3为载体的催化剂将难以应对更加严峻的挑战。为了进一步提高催化剂的加氢脱硫性能,大量技术人员研究了其他类型的催化剂载体材料,如Zhang等[4]通过水热法制备出钛硅沸石纳米棒(TS-1),负载钴、钼制备了CoMo/TS-1催化剂,经研究发现,TS-1骨架中的Ti物种可以增强Mo与TS-1间的相互作用,有助于暴露更多有利于加氢的活性位点,进而提高催化剂的加氢脱硫性能。Cortes-Jacome等[5]采用水热法制备出高比表面积的氧化钛纳米管,并以此为载体制备出高分散的CoMoS催化剂,可表现出良好的脱硫性能。另外,Huang等[6]以四正丁基钛酸为钛源,采用溶胶-凝胶法制备TiO2-Al2O3复合载体,负载镍、钼制备出具有高活性的加氢脱硫催化剂。
由上可见,Ti的引入能够有效避免纯γ-Al2O3载体催化剂上存在的问题,通过将TiO2与γ-Al2O3载体进行复合,可以有效地发挥TiO2与γ-Al2O3各自的优势,进而制备出优异的加氢脱硫催化剂。然而,常规TiO2作为载体存在比表面积小、机械强度差等缺点,虽然用溶胶-凝胶等[7-8]方法能够制备出高比表面积的TiO2,但是制备成本高、工业化困难等缺点限制了其在工业方面的应用,将其进一步与γ-Al2O3进行复合,会对所制得的复合载体的孔结构、两者间相互复合强度以及机械性能等因素产生负面影响。笔者所在课题组前期[9]从二钛酸钾(K2Ti2O5)晶须出发,成功制得比表面积大于 139 m2/g 的介孔TiO2晶须材料,所制得的TiO2成型载体抗破碎强度大于12 N/mm。刘金龙等[10]、朱银华等[11]、Li等[12]以TiO2为载体制备HDS催化剂,均显示出优异的催化活性。在此基础上,温广明等[13]开展介孔TiO2晶须与γ-Al2O3的复合制备研究,可以有效克服常规TiO2与γ-Al2O3复合所存在的不足,并发现介孔TiO2晶须在复合载体中的最佳质量分数为5%,初步负载活性组分后,所得催化剂可表现出优异的加氢脱硫性能。
然而,已有文献[14-15]对不同载体进行活性组分负载过程对比考察的结果表明,催化剂性能受到载体的表面性质、孔结构、晶相等多方面因素影响,所得到的实验结果难以直接借鉴到其他类型的催化剂载体,故针对新型载体材料需要重新开展活性组分负载研究。因此,笔者对新型的介孔TiO2晶须-γ-Al2O3复合载体开展活性组分负载量的系统性研究,通过一系列技术手段对所制备的催化剂进行结构表征,并以DBT作为模型反应物对催化剂的性能进行评价。
1 实验部分
1.1 试剂和原料
钼酸铵((NH4)6Mo7O24·4H2O)、二硫化碳(CS2)、十氢萘(C10H18),均为分析纯,国药集团化学试剂有限公司产品;二苯并噻吩(C12H8S,质量分数99%),麦克林试剂有限公司产品;硝酸(HNO3,质量分数69%),国药集团化学试剂有限公司产品。介孔TiO2晶须,自制,制备方法参考文献[7];拟薄水铝石,工业级,淄博万霖化工科技有限公司产品;TiO2(比表面积11.9 m2/g),分析纯,国药集团化学试剂有限公司产品。
1.2 载体和催化剂的制备
采用常规的溶胶-凝胶法制备介孔TiO2晶须-γ-Al2O3复合载体,具体步骤如下:称取10 g拟薄水铝石放入装有200 mL去离子水的三角烧瓶中,搅拌混匀;将三口烧瓶置于油浴锅中加热至70 ℃并保持恒温,缓慢滴加稀硝酸,将混合物体系pH值调至1左右,加入0.5 g介孔TiO2晶须;继续搅拌6 h,期间每隔0.5 h补充适量的稀硝酸维持体系pH=1;结束后,缓慢滴加适量氨水,将体系pH值调至9左右,形成复合胶状物,陈化3 h后取出,用去离子水将其洗至pH=7,经抽滤、烘干、550 ℃ 焙烧2 h后得到介孔TiO2晶须-γ-Al2O3复合载体,记为TiO2-Al2O3。以常规TiO2为钛源,采用同样的方法制备TiO2质量分数为5%的复合载体,记为c-TiO2-Al2O3。
以(NH4)6Mo7O24·4H2O作为钼源配制前驱体溶液,通过等体积浸渍法负载活性组分。将一定量前驱体溶液加入到载体中,经搅拌、陈化、干燥和500 ℃焙烧2 h后得到不同MoO3含量的介孔TiO2晶须-γ-Al2O3复合载体催化剂,记为x-MoO3/TiO2-Al2O3,其中x为MoO3的质量分数。另外,采用同样的方法制备纯γ-Al2O3为载体及c-TiO2-Al2O3为载体MoO3质量分数为20%的催化剂作为参比样,分别记为20%MoO3/γ-Al2O3和20%MoO3/c-TiO2-Al2O3。
1.3 催化剂表征
采用美国Micromeritics公司TristarII 3020M型比表面孔隙吸附仪分析各个催化剂的孔结构,测试前,样品在150 ℃下脱气处理6 h,以N2作为吸附质在液氮(-196 ℃)下进行测定。样品的微观形貌结构分析以及EDX测试采用日立公司生产的Hitachi S-4800场发射扫描电镜(FESEM)进行,操作电压为5 kV。硫化后催化剂的形貌分析在JEOL JEM-2100透射电子显微镜上进行,通过TEM显微照片确定的颗粒尺寸来统计评估MoS2颗粒的尺寸信息。样品的晶体结构分析采用德国Bruke公司的D8 Adavance型X射线衍射仪,CuKα辐射源,管电流为100 mA,管电压为40 kV,扫描范围2θ=5°~80°,扫描步长为0.02°,扫描速率为20 s/step。样品的拉曼光谱分析在配备CCD相机探测器的Horiba HR 800光谱仪上进行,采用波长为 514 nm Ar/Kr离子激光器作为激发光源,物镜放大倍数为50倍,样品激光功率不超过5 mV。氢气程序升温还原(H2-TPR)实验在天津先权工贸发展有限公司的TP-5000型多用吸附仪上进行。硫化状态催化剂的X-射线光电子能谱分析在ESCALAB 250光谱仪(美国Thermo Fisher Scientific公司产品)上进行,AlKαX射线激发源,功率300 W。
1.4 催化剂活性评价
催化剂的加氢脱硫性能在固定床微反应装置上进行评价。将0.33 g催化剂放置在反应器的恒温段,反应前将催化剂置于3%(质量分数)CS2/十氢萘溶液中,在温度300 ℃、H2压力2 MPa、H2流量20 mL/min、硫化液流量0.033 mL/min条件下预硫化处理6 h。预硫化结束后,将硫化液切换成1%(质量分数)DBT/十氢萘反应液,在温度300 ℃、H2压力2 MPa、氢/油体积比600、体积空速6 h-1的条件下进行反应。预反应5 h稳定后开始采集第一个产物,之后每隔1 h采集一次反应产物。反应产物采用山东鲁南瑞虹化工仪器有限公司SP-6890 型气相色谱仪进行检测。DBT转化率通过公式(1)计算。
(1)
DBT的加氢脱硫途径主要有两种[12]:直接脱硫(DDS)产物为联苯(BP),氢化脱硫(HYD)产物为环己基苯(CHB)。DDS选择性根据公式(2)计算。
(2)
式(1)和(2)中,win和wout分别为原料和产物中DBT的质量分数,%;w为产物中联苯的质量分数,%。
2 结果与讨论
2.1 催化剂的物性表征结果
2.1.1 XRD分析
图1是TiO2-Al2O3复合载体和不同MoO3含量MoO3/TiO2-Al2O3催化剂以及20%MoO3/c-TiO2-Al2O3催化剂的XRD谱图。由图1可见,TiO2-Al2O3复合载体样品在2θ为46.0°和67.0°处的衍射峰对应于γ-Al2O3相[16]的特征峰,而在2θ为25.3°和48.0°两处微弱的衍射峰对应于锐钛矿相TiO2[17]的特征峰。此外,当MoO3负载质量分数为25%和30%时,在2θ为23.5°处出现明显的MoO3衍射峰[11],这主要是由于MoO3负载量过高,在载体表面发生了团聚所引起。当MoO3负载质量分数低于20%时,在MoO3/TiO2-Al2O3催化剂的XRD曲线上没有出现MoO3衍射峰,表明在该范围内活性金属在载体表面具有良好的分散性,这有利于催化剂还原和提高反应活性。而20%MoO3/c-TiO2-Al2O3催化剂在2θ为25.3°、37.7°、48.0°、53.8°和55.1°处均出现明显的锐钛矿型TiO2的衍射峰[17],并且在2θ为23.5°处出现明显的MoO3衍射峰。此结果表明20%MoO3/c-TiO2-Al2O3催化剂中的活性金属发生团聚,生成了晶态MoO3,而MoO3负载质量分数为20%时,20%MoO3/TiO2-Al2O3催化剂中并没有出现晶态MoO3的衍射峰。
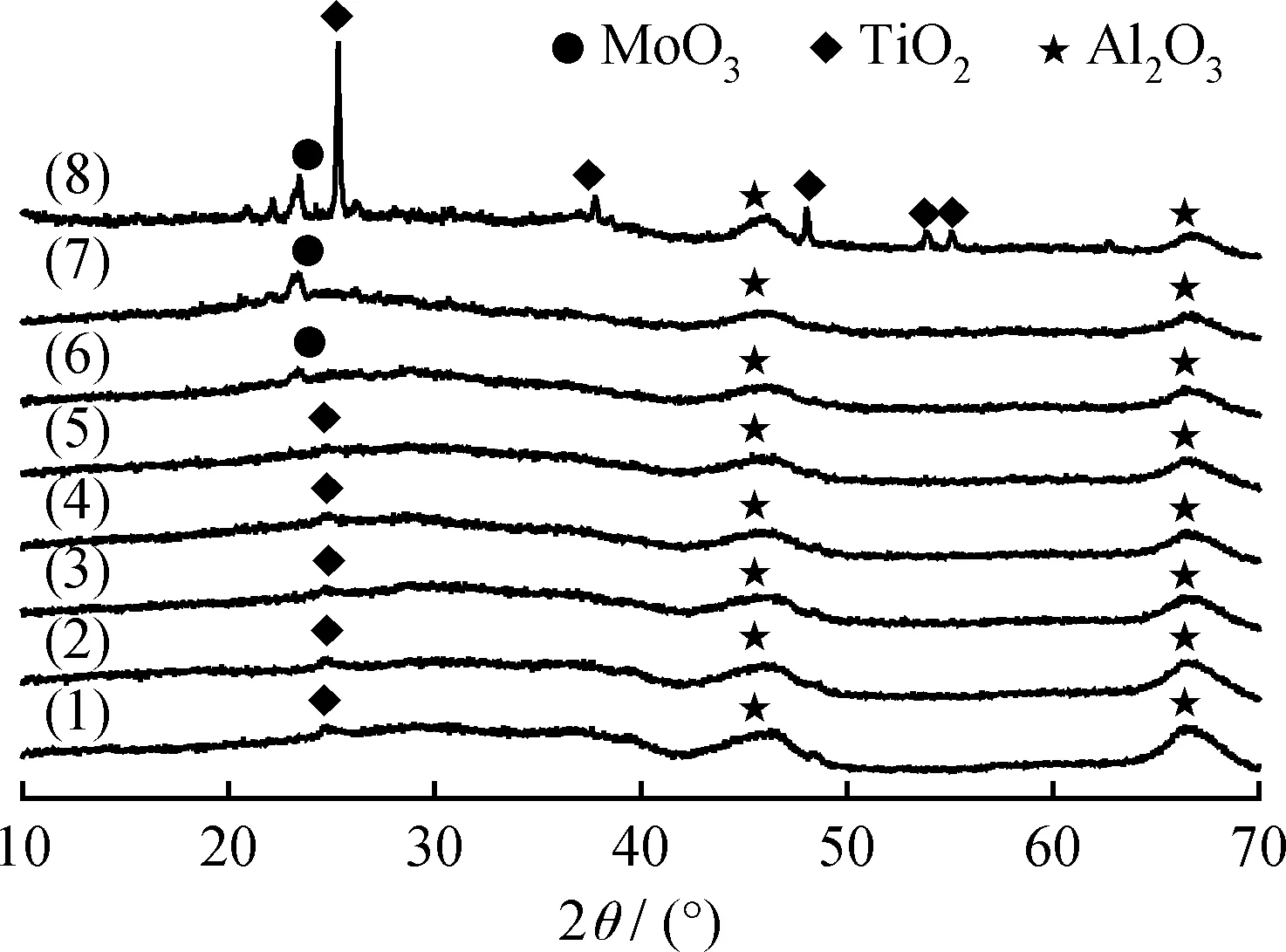
图1 TiO2-Al2O3复合载体以及负载不同含量MoO3的催化剂XRD图谱
2.1.2 比表面积和孔结构分析
图2为TiO2-Al2O3复合载体以及负载不同含量MoO3的催化剂的N2吸附-脱附等温线。由图2(a)可见,所有样品的N2吸附-脱附等温线均呈IV型,这表明所有材料均具有介孔结构[18]。图2(b)的孔径分布图显示,各催化剂以及复合载体的最可几孔径主要集中在5 nm左右,进一步证明复合载体催化剂具有介孔结构。
表1为复合载体及其不同MoO3含量催化剂的比表面积、平均孔径及孔体积数据。由表1可知,TiO2-Al2O3复合载体的比表面积为308.7 m2/g,孔体积为0.56 cm3/g,平均孔径为6.1 nm,具有丰富的孔结构。而常规TiO2为Ti源的c-TiO2-Al2O3复合载体的比表面积仅为203.4 m2/g,孔体积为0.32 cm3/g,平均孔径为4.5 nm。负载活性组分MoO3后,所制得的催化剂比表面积和孔体积均有所降低,当MoO3负载质量分数为30%时,MoO3/TiO2-Al2O3催化剂的比表面积和孔体积最低;而20%MoO3/c-TiO2-Al2O3催化剂的比表面积远小于20%MoO3/TiO2-Al2O3的。负载MoO3后催化剂的比表面积和孔结构数据减小是由于金属负载于载体表面和孔道之中,导致载体部分表面以及小孔孔道被覆盖或堵塞[19]。
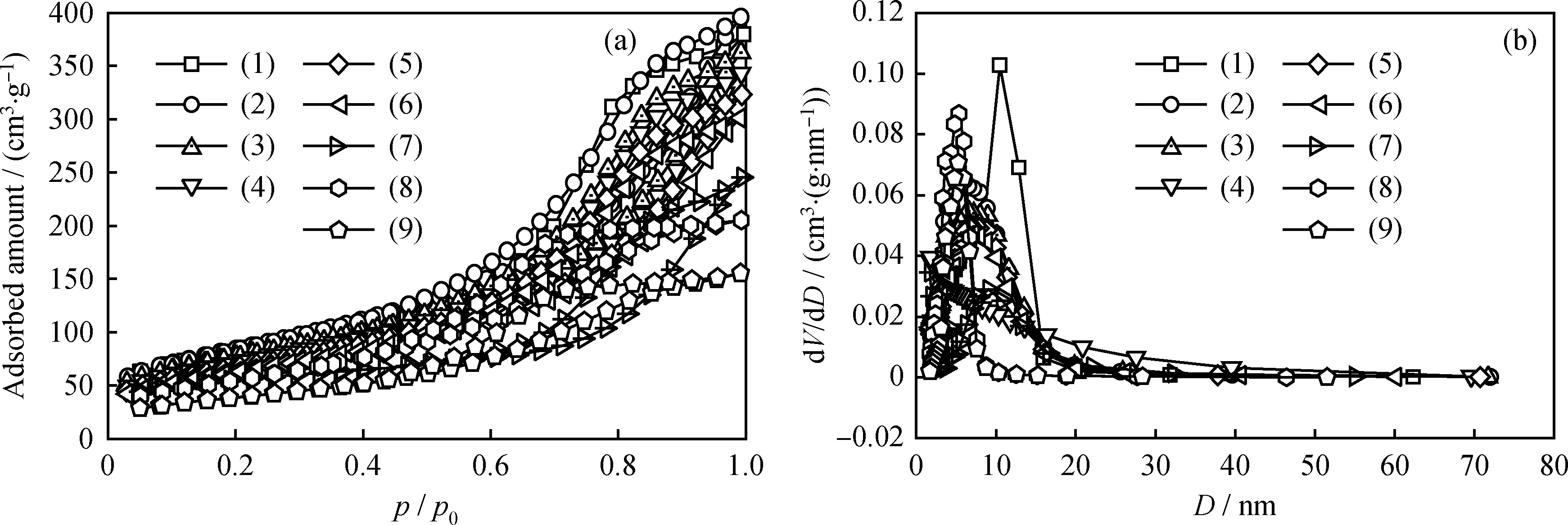
图2 TiO2-Al2O3复合载体与不同MoO3含量催化剂的N2吸附-脱附曲线和孔径分布图
2.1.3 Raman分析
图3给出了不同MoO3负载量的MoO3/TiO2-Al2O3复合载体催化剂的Raman光谱图。由图3可知:所有催化剂在640 cm-1处出现的Raman信号峰对应于锐钛矿型TiO2的特征峰[20];在865 cm-1处出现的Raman信号峰与低聚体MoOx中桥接Mo—O—Mo的对称和反对称伸缩振动有关[21],并且随着催化剂中Mo含量增加,拉曼振动峰强度先增加后减少;在930~970 cm-1范围内的宽信号峰可能与氧化钼中的Mo=O的伸缩振动有关[22-23],当MoO3负载量较低时,930~970 cm-1几乎没有拉曼峰出现,随着MoO3负载量增加,930~970 cm-1的拉曼信号峰强度明显增加,表明在催化剂表面形成了更多 Mo=O 的Mo物种,有报道指出这种Mo物种有利于提高催化剂活性[24]。
2.1.4 SEM和TEM分析
图4是5%MoO3/TiO2-Al2O3、20%MoO3/TiO2-Al2O3和30%MoO3/TiO2-Al2O3催化剂的SEM照片。由图4可以看出,金属负载量的增加并没有对载体的形貌产生大的影响。为了证明氧化状态的Mo在载体表面的分散度差异,采用SEM-EDX对 20%MoO3/TiO2-Al2O3和30%MoO3/TiO2-Al2O32个催化剂进行EDX测试,在每个催化剂中随机取2点进行测试,结果如图5所示。由图5可知,MoO3
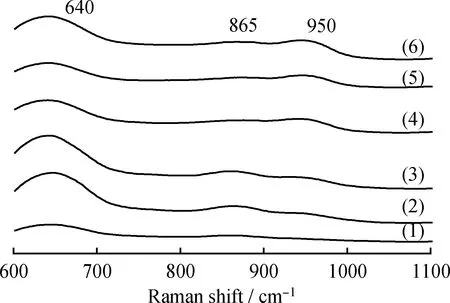
图3 不同MoO3负载量的MoO3/TiO2-Al2O3复合载体催化剂Raman光谱图
负载质量分数为20%时,催化剂表面所测得的Mo金属元素的质量分数分别为13.17%和13.04%,Mo含量基本一致,表明MoO3质量分数为20%时在载体表面分散均匀;MoO3负载质量分数为30%时,催化剂表面所测得的Mo元素的质量分数分别为18.12%和19.51%,差别较大,此结果表明,MoO3质量分数为30%时,活性金属在载体表面的团簇使局部Mo金属含量偏高。图6是新鲜硫化的20%MoO3/TiO2-Al2O3和30%MoO3/TiO2-Al2O3催化剂的TEM照片。TEM结果表明各催化剂表面的MoS2均呈现层状结构。通过统计(对150个MoS2进行尺寸统计)和计算[25]得到20%MoO3/TiO2-Al2O3和30%MoO3/TiO2-Al2O3的MoS2片晶的平均长度分别为2.5 nm和2.7 nm,堆叠层数分别为1.9和2.0。
2.1.5 H2-TPR分析
图7是不同MoO3含量MoO3/TiO2-Al2O3催化剂的H2-TPR图谱。由图7看到,所有催化剂的TPR曲线均在350~500 ℃范围内出现1个明显的还原峰,这对应于八面体配位的聚合Mo物种从Mo6+到Mo4+的还原过程。Zhou等认为,该类型的Mo物种与载体之间的相互作用弱,易于还原成具有优异加氢脱硫性能的活性物种[26]。MoO3负载质量分数低于20%时,催化剂还原峰峰值为475 ℃,当MoO3负载质量分数高于20%时,催化剂的还原温度略有升高,还原峰的峰值由475 ℃升高至494 ℃,并且在500~600 ℃范围内出现1个小的还

图4 不同MoO3含量MoO3/TiO2-Al2O3催化剂的SEM照片
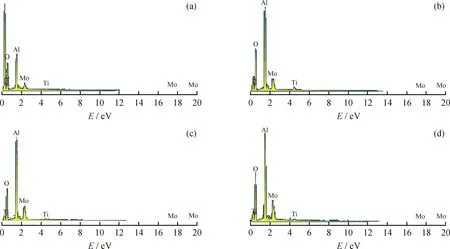
图5 不同MoO3含量MoO3/TiO2-Al2O3催化剂的SEM-EDX谱

图6 不同MoO3含量MoO3/TiO2-Al2O3催化剂的TEM图
原峰。据报道,晶态MoO3在480、575和665 ℃处出现还原峰[27],而图1的XRD结果显示,MoO3负载质量分数超过20%时催化剂上会出现晶态MoO3,由此判断该还原峰为晶态MoO3的还原峰。图7中还看到,20%MoO3/γ-Al2O3催化剂的还原峰峰值在506 ℃,而MoO3/TiO2-Al2O3催化剂的还原峰峰值明显低于506 ℃,表明TiO2的引入能够有效减弱Mo与Al2O3之间的相互作用,从而促进形成更多的活性相。
2.1.6 XPS分析
图8是硫化态的20%MoO3/TiO2-Al2O3催化剂的Mo 3d的XPS图谱。由图8可知,硫化后的催化剂主要存在2种价态的Mo物种,特征峰的结合能位置分别为232.5和238.6 eV,可归属于Mo6+物种[28](Mo VI: (232.5±0.1) eV 3d5/2, (238.6±0.1) eV 3d3/2),特征峰的结合能位置分别为229 eV和232.1 eV的可归因于Mo4+物种(Mo IV:(229.0±0.1) eV 3d5/2, (232.2±0.1) eV 3d3/2)[28]。van Haandel等[29]对比研究了硫化前后的Mo物种结合能变化情况,发现硫化前催化剂中的Mo物种只在高结合能位置(232.5 eV和238.6 eV)出现特征峰,经硫化后,Mo物种在高结合能位置和低结合能(229.0 eV和232.3 eV)位置均出现特征峰,认为这是催化剂中高价态的Mo物种被还原成低价态Mo物种所致。同理,图8中在低结合能处出现的Mo4+物种信号表明催化剂中Mo物种在硫化过程中发生还原。通过分峰计算可知20%MoO3/TiO2-Al2O3催化剂的硫化度为32.2%(硫化度计算公式:F=AMo4+/(AMo4++AMo6+)×100%,其中F为硫化度,AMo4+和AMo6+分别代表XPS中Mo4+和Mo6+的特征峰面积),Zhang等[30]在相同硫化条件下发现MoO3/Al2O3系列催化剂中Mo的硫化度为22%~31%,显然,本实验中MoO3/TiO2-Al2O3催化剂的硫化度更高,这可归因于MoO3/TiO2-Al2O3催化剂中的TiO2促进了MoO3发生硫化。
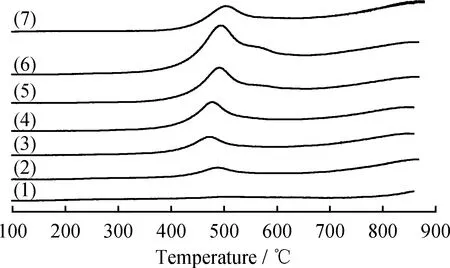
图7 不同MoO3负载量的MoO3/TiO2-Al2O3复合载体催化剂和20%MoO3/γ-Al2O3的H2-TPR谱图
2.2 不同催化剂样品的DBT加氢脱硫反应性能评价
不同催化剂的DBT加氢脱硫反应结果如表2所示。表2表明:MoO3/TiO2-Al2O3催化剂的DBT转化率随MoO3负载量增加呈先增加后降低的趋势,当MoO3负载质量分数为20%时,MoO3/TiO2-Al2O3催化剂在当前的反应条件下表现出最佳的加氢脱硫性能,其DBT转化率达到56%;20%MoO3/γ-Al2O3和20%MoO3/c-TiO2-Al2O3的DBT转化率分别为49%和41%,由此可见介孔TiO2晶须-Al2O3复合载体作为脱硫催化剂载体可以表现出比纯γ-Al2O3和常规TiO2为Ti源的复合载体更为优异的深度加氢脱硫性能。Ferdous等[31]报道,Ti的引入可以有效促进Mo在Al2O3催化剂表面形成更多活性多钼氧化物。因此可以解释介孔TiO2晶须-Al2O3复合载体催化剂的脱硫性能优于γ-Al2O3催化剂。常规TiO2为Ti源的c-TiO2-Al2O3载体催化剂的加氢脱硫活性低于介孔TiO2晶须-Al2O3复合载体催化剂和γ-Al2O3催化剂,可能是由于载体比表面积低,使得Mo金属在载体表面团聚形成不利于催化活性的晶态MoO3。而另一方面,由2.1节表征结果可知,当MoO3负载量较低时,MoO3能够在载体表面良好地分散,在该条件下,催化剂的活性位点数量随负载量增加也得到了进一步的提升;然而,当负载质量分数高于分散阈值(20%)时,MoO3在载体的分散度有所下降,活性金属团聚形成了难以还原的晶态MoO3,进而使得催化剂的DBT转化率下降。
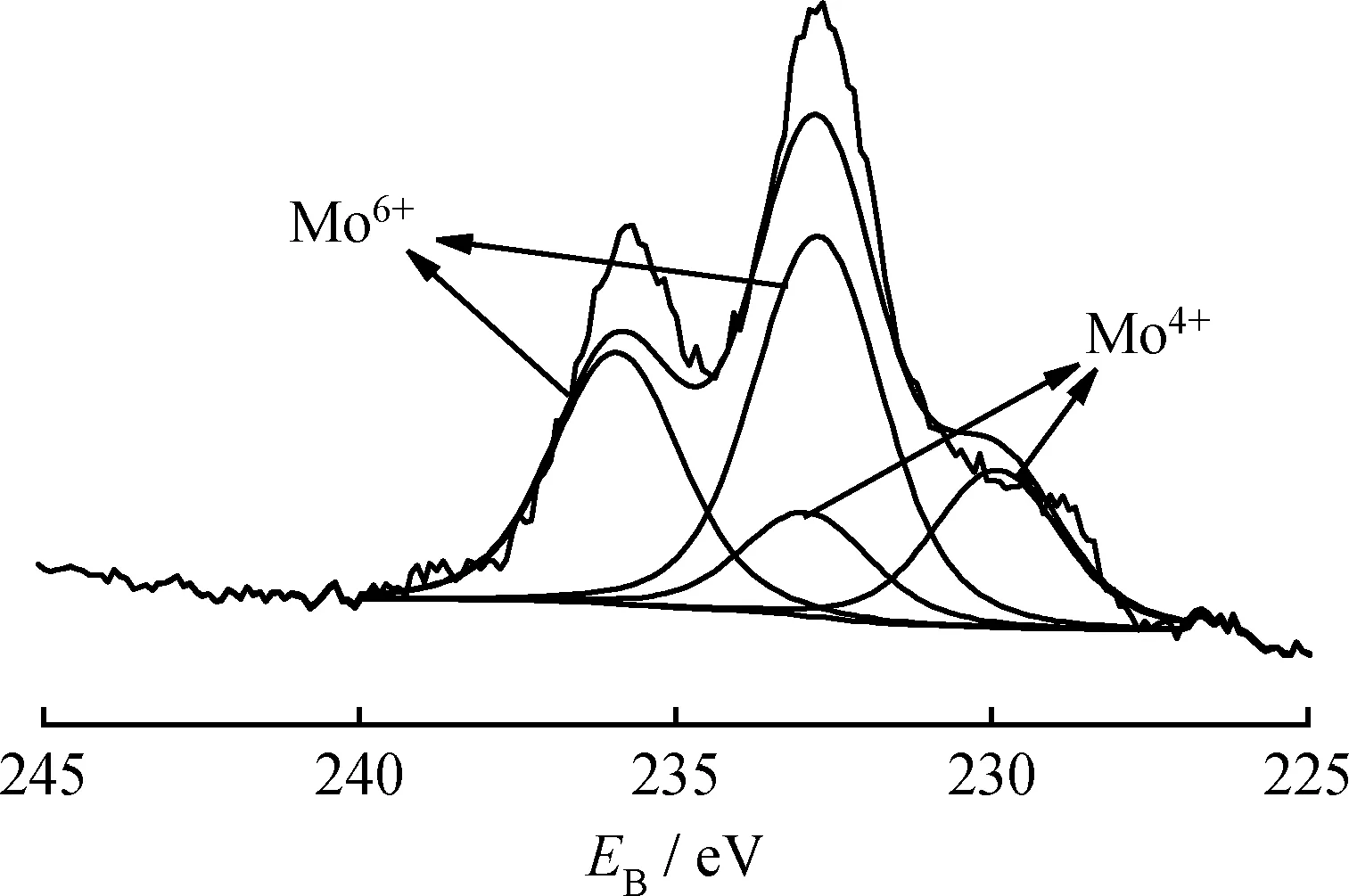
图8 硫化后20%MoO3/TiO2-Al2O3催化剂的XPS谱图
由表2可知,不同MoO3负载量的 MoO3/TiO2-Al2O3催化剂的DDS选择性均在70%左右,表明MoO3负载量的变化没有对催化剂的DDS选择性产生明显的影响。20%MoO3/c-TiO2-Al2O3催化剂的DDS选择性在57%左右,而20%MoO3/γ-Al2O3催化剂的DDS选择性仅为46%,明显低于复合载体催化剂,表明Ti引入使得催化剂更加倾向于DDS路径。Vazquez-Garrido等[32]和Guevara等[33]的研究结果同样表明,TiO2能够有效减弱活性金属与Al2O3间相互作用,从而增加催化剂的DDS选择性。
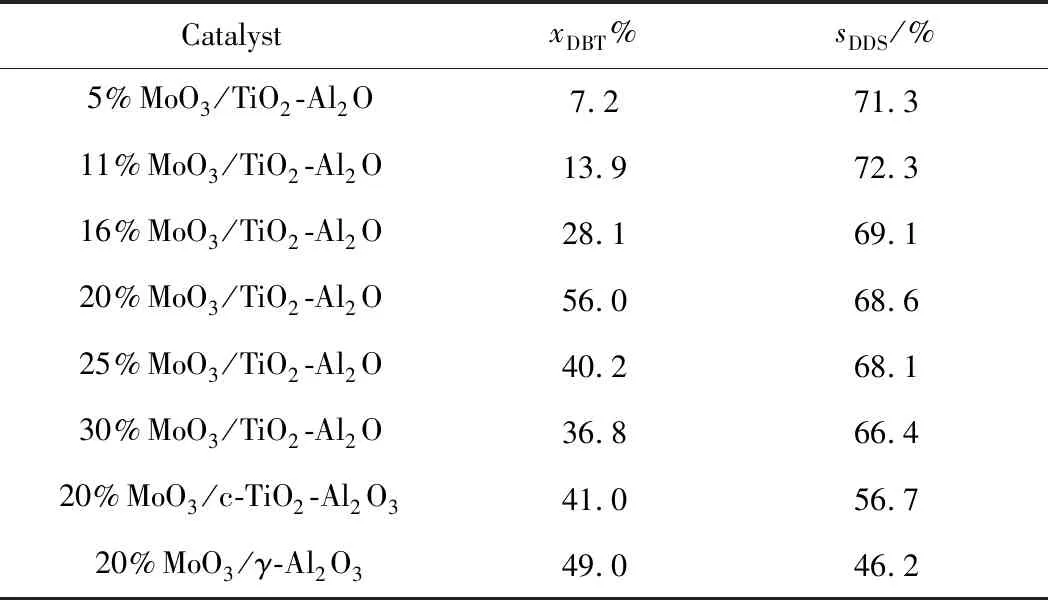
表2 不同催化剂加氢脱硫反应的DBT转化率和DDS选择性
3 结 论
制备了介孔TiO2晶须与γ-Al2O3复合载体(TiO2-Al2O3)及不同MoO3负载量的MoO3/TiO2-Al2O3催化剂,并进行了物性表征和对二苯并噻吩的加氢脱硫反应性能评价,发现当MoO3负载质量分数为20%时,MoO3在复合载体上分散良好,所制得的20%MoO3/TiO2-Al2O催化剂具有高的比表面积和丰富的介孔结构,活性组分易于还原,能够展示出最佳的加氢脱硫性能,DBT转化率达到56%,优于相同条件下纯γ-Al2O3为载体的催化剂(49%)。
猜你喜欢
石油化工高等学校学报(2022年1期)2022-04-15
陶瓷学报(2021年5期)2021-11-22
烟台果树(2021年3期)2021-07-21
建材发展导向(2021年7期)2021-07-16
陶瓷学报(2020年6期)2021-01-26
上海电力大学学报(2020年6期)2020-12-25
燃料化学学报(2019年10期)2019-11-04
中外葡萄与葡萄酒(2019年2期)2019-04-12
上海农业科技(2019年1期)2019-02-22
人工晶体学报(2019年1期)2019-02-19
