
Hunting down the dominating subclone of cancer stem cells as a potential new therapeutic target in multiple myeloma:An artificial intelligence perspective
2020-09-18 13:27
World Journal of Stem Cells 2020年8期
Lisa X Lee,Division of Hematology/Oncology,Department of Medicine,Chao Family Comprehensive Cancer Center,UCI Health,Orange,CA 92868,United States
Shengwen Calvin Li,Neuro-oncology and Stem Cell Research Laboratory,CHOC Children's Research Institute,Children's Hospital of Orange County,Orange,CA 92868,United States
Shengwen Calvin Li,Department of Neurology,University of California-Irvine School of Medicine,Orange,CA 92868,United States
Abstract
Key words:Multiple myeloma;Single cells;Single-cell transcriptome;Subclonal evolution;Cancer stem cells;Systemic tracking of single-cell landscape;Artificial intelligence medicine
INTRODUCTION
Epidemiology
With approximately 31000 new cases of multiple myeloma (MM) diagnosed in the United States (US) per year,the impact of this incurable disease on individual patients and society as a whole is profound.The median age at diagnosis is 70 years old[1].All diagnoses of MM are believed to be preceded by a state of clonal expansion of plasma cells (PCs),including monoclonal gammopathy of unknown significance (MGUS) and smoldering myeloma (SM).The duration of these precursor conditions of MM has been demonstrated to be present up to 15 years prior to the diagnosis of MM[2].
Diagnosis and disease prognostication
The current diagnosis of MM requires a bone marrow biopsy and aspirate,which is used to enumerate plasma cell content and to characterize PCs by immunohistochemical staining,cytogenetics,and flow cytometry.Detection of cytogenetic alterations,in particular,are paramount to provide prognostication and direct therapy and have been incorporated into the standardized staging system for MM[3].For example,the presence of high-risk cytogenetics,including del17p,t(4,14),and t(14;16) prognosticates for survival 1/5ththat of standard-risk cytogenetics[4].However,the identification of such cytogenetic features may be used to guide therapy such as in patients with t(4;14),who have traditionally had significantly inferior outcome may be able to have an overall survival (OS) similar to patients with standard-risk MM when treated with bortezomib-containing regimens and autologous stem cell transplantation[5].
CURRENT SOLUTIONS TO OVERCOME THERAPEUTIC RESISTANCE
Initial treatment incorporating conventional drugs such as Dexamethasone (Dex)effectively induces MM cell death;however,prolonged drug exposures result in the development of chemoresistance.Thus,individual patients' survival within a risk category remains variable,and the patients relapse despite achieving a “complete response,” reflecting persistent disease that cannot be detected using the currently recommended disease evaluation techniques.It is becoming apparent that static cytogenetic categories alone are not sufficient to define subclone formation and stage[6].Several methods are being evaluated to enhance further our ability to individualize treatment.
First,response assessment using minimal residual disease (MRD) at varying time points in a patient's disease process can further fine-tune response-adapted treatment strategies.MRD negativity at any given time point is closely correlated with more prolonged progression-free survival (PFS).It has been incorporated into the International Myeloma Working Group recommendation for response assessment,and ongoing studies are studying adaptive treatment strategies based on achieving MRD negativity[7].Current methods for minimal residual disease testing include flow cytometry or next-generation sequencing.Multiparametric flow cytometry (MFC)MRD testing in MM has quite low sensitivity,detecting one cell in 104cells and requires at least 2 × 106,preferably greater than 5 × 106bone marrow cells to be measured[8],as recommended by the International Clinical Cytometry Society and the European Society for Clinical Cell Analysis.In addition to low sensitivity,MFC may be unable to differentiate between the dominant clone and various subclones[9].In addition,there is heterogeneity between laboratories (cross-platform flow cytometry),which depends on instrumentation used and initial gating parameters (CD38,CD138,CD45,forward,and sideward light scatter)[10],within the same aliquot and is therefore entirely subjective.Next-generation sequencing (NGS) of immunoglobulin gene sequences is an alternative method for MRD assessment.While a more sensitive technique compared with MFC,detecting one tumor cell in 106,NGS cannot detect mutations that are present within individual cells[11].Therefore,based on our current technologies for MRD detection,we can only say whether a patient is positive or negative without genuinely understanding the temporal and spatial heterogeneity within a given plasma cell population.
Second,we recognize the temporal and spatial heterogeneity in MM,as clinical observations revealed that several subclones of PCs exist at diagnosis and that there is selective therapeutic pressure for the evolution of individual subclones.This phenomenon can be tracked utilizing the whole-genome sequencing of paired tumor/normal samples.In one study,from 203 MM patients revealed frequent mutations in KRAS,NRAS,BRAF,FAM46C,TP53,and DIS3.Mutations were often present in subclonal populations,and multiple mutations within the same pathway (e.g.,KRAS,NRAS,and BRAF) were observed in the same patient"[12].However,a more recent study utilizing plasma samples found mutations in the KRAS-MAPK pathway in 70% of samples in addition to multiple mutations within subclones including a notable mutation in PIK3CA[13]signifying perhaps the relative insensitivity of a one site biopsy in addition to the development of more mutated clones with the escape of PCs from the bone marrow microenvironment.Liquid biopsy utilizing cfDNA can provide us with information on targetable mutations,but is a way to study spatial and temporal heterogeneity present.The drawbacks for above current testing mistaken population phenomena for real physiological events happening only within a singlecell (i.e.,subclone) – mutations exist in different cells may not cross-talk - thereby not being able to manifest as clinically treatable phenotypes – which would not give early insight into the evolution of a given patient's MM[14].
Third,the identification of chemoresistant biomarkers offers a trace to the subclones,e.g.,the oligonucleotide array analysis demonstrates that heat shock protein-27 (Hsp27) is upregulated in Dex-resistant,but not in Dex-sensitive MM cells.Proteomics analysis of Hsp27-immunocomplexes revealed the presence of actin in Dex-resistant,but not in Dex-sensitive cells.The activator protein-1 transcription factor family (JUNB) driving the JunB-mediated phenotype in MM cells:knockdown of JUNB restored the response to dexamethasone in dexamethasone-resistant MM cells.When JunB-ER fusion protein in dexamethasone-sensitive MM cells is activated by 4-hydroxytamoxifen,Dex-sensitive cells become to be resistant to dexamethasone- and bortezomib-induced cytotoxicity[15].
Thus,the ability to track mutations within a single cell subclone lends to the study of mechanisms of drug resistance,possibly leading to a better selection of targeted therapies.To that end,new technology must be developed and raised its sensitivity sufficient to evaluate the low burden of MM cells,which is currently being investigated as a way to detect pre-biochemical relapse[16].We propose to develop a technique that combines the detection of low-frequency events combines with the indepth characterization of the remaining subclones.
DEVELOPMENT OF AN INNOVATIVE SINGLE-CELL MOLECULAR PROFILING PLATFORM
While single-cell proteomics is still uncertain,single-cell RNA-seq is a widespread practice in research laboratories now.Many microfluidic devices,including ours,have been developed for single-cell transcriptome analysis[17]but clinical application of single-cell transcriptome is still not common,especially in cancer characterization and classification.US Food and Drug Administration approved CellSearch™ (Janssen,Raritan,NJ) of circulating tumor cells (CTC) for predicting PFS and OS in metastatic breast cancer for clinical use in 2005.However,CellSearch®data correlate negatively with survival in patients with metastatic breast,colorectal,or prostate cancer[18].CellSearch™,along with several other CTC enrichment techniques,relies on only fluorescent imaging analysis.
At least 50 competitor circulating tumor cell platforms exist (Table1)[19,20],only one,“CellSearch™” (Janssen,Raritan,NJ) was cleared by the US Food and Drug Administration in October 2005.As we have a prototype Multi-Phase Laser-cavitation Single Cell Analyzer (MLSCA) device for single-cell transcriptome analysis (Figure1),we develop a necessary commercialization 510k,and the CAP/CLIA component of this proposal must be side by side comparison of the applicant's technology to the CellSearch™ technology.This feature differentiates it from numerous competitor platforms used for single-cell counting.CellSearch™ can be used for predicting PFS and OS in metastatic breast cancer[19],however;CellSearch™ cannot generate consistent results for routine clinical use[21],due to the limit of the sensitivity of these devices[22].CellSearch™ captures CTCs from blood using magnetic particles coated with anti-EpCAM (CD326;17-1A antigen) antibodies[23],which relies on an antibody that binds to the protein EpCAM (epithelial cell adhesion molecule),present on the surface of malignant epithelial cells but not of blood cells.As CellSearch®,along with several other CTC enrichment techniques,relies on the presence of epithelial cell markers,CTCs that do not express EpCAM,such as those that have undergone an epithelial to mesenchymal transition may be missed[24,25].
Thus,CellSearch™ cannot generate consistent results for routine clinical use[21],and there is an unmet need to combine image analysis with RNA-seq for cancer classification.To fill this unmet need,we developed a microfluidic prototype device to connect FACS and imaging analysis (FISH) with molecular analysis (e.g.,single-cell transcriptomes) - whose prototype device is called MLSCA (Figure1).
Our MLSCA can overcome the limitation of “CellSearch™" [multiple-step processing,i.e.,separated reverse transcriptase polymerase chain reaction (RT-PCR)],and the strength is the ability of our MLSCA to perform RT-PCR on a single cell isolated on the chip (one-step processing),eliminating process errors.Our microfluidic system is equipped with both single-cell isolation and cDNA synthesis capabilities.Thus,our MLSCA enables (1) microscale fluorescence-activated cell selection for separation of rare cell subpopulations;and (2) generation of the high-quality singlecell transcriptome with nano-droplets.We have conducted and published a small Phase I clinical trial with our devices in myeloma risk stratification of MM[26].As a proof-of-concept for instrumentation of our prototype device,we published the clinical trial using MLSCA/MF-CD45-TACs on MM 48 patients[17],which shed new light for scale-up applications in clinics.
Conventional single-cell isolation techniques with microliter carry-over volumes cannot be used for sensitive nano-liter RT directly.Unlike PCR,which is a repetitive event that itself may introduce bias,RT is a single biochemical reaction for which starting mRNA concentration is critical.Further,the resultant cDNA population is thought to be unbiased.The innovations of our device include:(1) The single cell analyzer (MLSCA) combines single-cell fluorescent-activated cell selection,reverse transcript synthesis of high-quality cDNA,transcriptome analysis - all in 0.1 nano-liter droplets (50 pico-liter),thereby reducing procedure errors to improve single-cell cDNA quality and to yield reliable single-cell transcriptomes down-stream;(2) The ability to create single artificial cells,a highly controllable test system,for evaluation of platform performance,using homogeneous droplets of known low abundant RNA;(3)Demonstration of such MLSCA analysis clinical specimens;(4) Development of statistical methods for defining and quantifying molecular heterogeneity in populations of cells;and (5) Linkage to an RNAseq platform (Helicos and Ion Torrent)that does not require PCR amplification of the input cDNA is crucial for ensuring the fidelity of the measured transcriptome:read counts can be directly related to RNA abundance,with no possible distortion due to differential PCR efficiencies.Thus,the strength is the ability of the MLSCA to perform RT-PCR on a single cell on the chip.This strength differentiates it from numerous competitor platforms only used for CTC counting.We tested the MLSCA by addressing MM heterogeneity to identify underlying tumor initiation,and relapse biomarkers in MM,which was inspired by that fact that single-cell transcriptomic analysis in medulloblastomas led to mapping oncogenic networks including HIPPO-YAP/TAZ and AURORA-A/MYCNpathways[27]and to identify pathways of drug resistance[28].
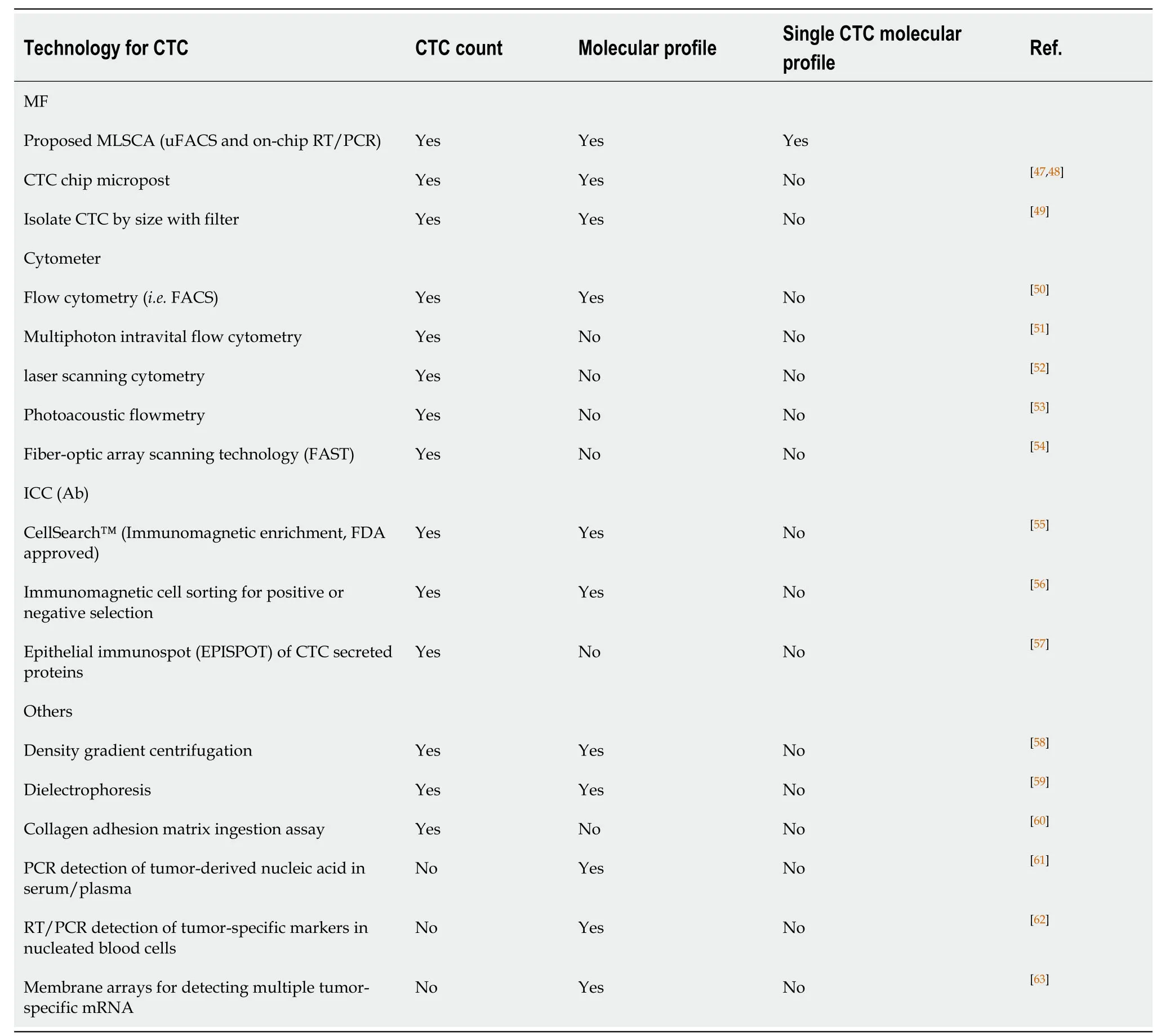
Table1 Multi-Phase Laser-cavitation Single Cell Analyzer can perform both circulating tumor cells enumeration and single-cell molecular characterization
We applied this MLSCA for a clinical trial of risk stratification of MM[17].As our MFCD45-TACs differs from CellSearch™,we expected single-cell features (FACS,biomarkers) of CellSearch™ differ from those of our MF-CD45-TACs (Figure2).Specifically,our MF-CD45-TACs-based technology distinguished CD45-cells from MM PCs,which improved the detection of rare genetic alternation in PCs,which was a significant improvement over direct flow cytometry and FISH,and led to more precise diagnosis and prognosis of MM.
This attribute is of significance as MM is an incurable neoplasm of PCs that affects more than 20000 people annually in the United States.Risk stratification,primarily based on cytogenetic abnormalities,has emerged as essential for its management[29].Thalidomide,lenalidomide,and pomalidomide,first to the third generation of immunomodulatory drugs (IMiDs),respectively,are used for maintenance therapy of MM.Cytogenetic alterations are the base of risk stratification for MM and the selection of which IMiDs for MM therapy[30].

Figure1 A prototype LSCAT device for single-cell transcriptome analysis.
The rarity and sporadic distribution of PCs in bone marrow often lead to falsenegative results of FACS and cytogenetic detection performed directly on a bone marrow biopsy sample.Target cell enrichment could overcome the rarity and sporadic distribution of PCs in the bone marrow.Density gradient centrifugation and magnetically labeled antibodies [DG/magnetic-activated cell sorting (MACS)] with MACS have been widely used to enrich target cells in blood samples.MACS enrichment of CD138+cells for FISH in MM diagnosis has been reported[31].However,MM cells with low levels of CD138 have also been associated with poor prognosis[32].Therefore,a better enrichment method is needed.Here,we report a novel microfluidic approach,combining microfluidic size selection and CD45 depletion with tetrameric antibody complexes (TACs) for the enrichment of MM cells (MF-CD45-TACs) in bone marrow samples.Our study showed that this approach significantly improves the detection of rare genetic alternations in PCs.Parallel diagnosis performed for 48 patients (Figure3) showed that the microfluidic enrichment approach represents a significant improvement over direct flow cytometry and FISH and leads to more precise diagnosis and prognosis[17].Implementation of this modified diagnostic assay in clinics could improve the current clinical outcomes of MM (Figure4 and Figure5).
APPLICATION OF SINGLE CELL SUBCLONE TRACKING TO THE FUTURE TREATMENT OF MM
At the MGUS/Smoldering stage
While all MM is preceded by an asymptomatic MGUS/smoldering myeloma stage[33],only a fraction of these individuals will evolve to symptomatic MM.Currently,some high-risk features such as high bone-marrow plasma cell burden,light chain ratios,and predicates the development of symptomatic MM.Still,we do not understand the oncogenesis of MM and,therefore,cannot accurately determine who will progress and who will remain asymptomatic.Using circulating tumor cell technology,we could track the occurrence of trigger genetic events in pre-symptomatic patients without the need to perform repeated invasive procedures and potentially intervene to eradicate these emerging malignant subclones using targeted therapies prior to the development of symptomatic disease or the acquisition of additional potent genetic mutations.
MRD monitoring
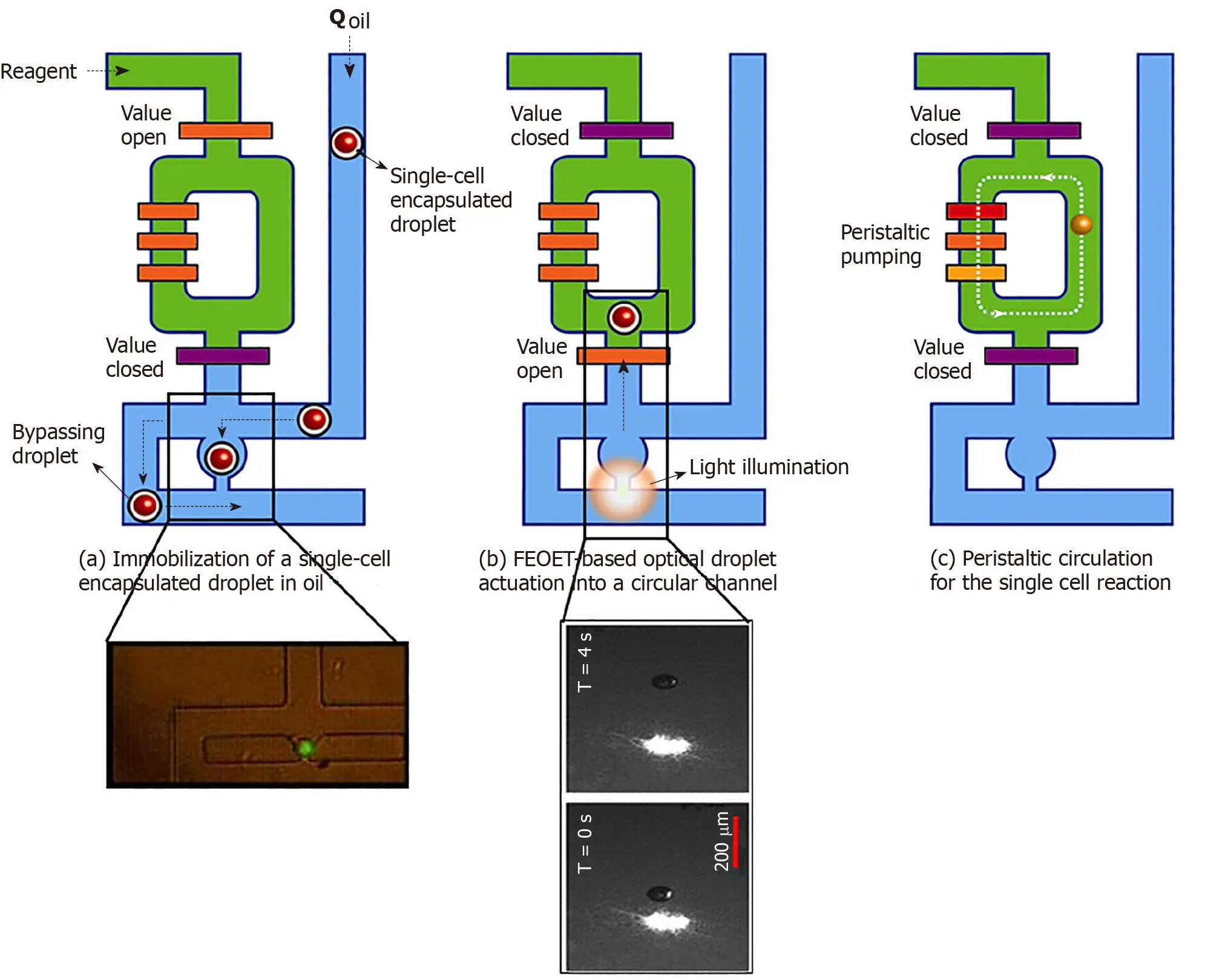
Figure2 Processing of single-cell droplets.A:A single-cell in an oil droplet travels to a trapping module of the 10-nl reactor (green,in inset).Other cells are forced to bypass to the next unit;B:FEOET push the droplet (with a cell) past the open valve (orange bar),which closes,locking the droplet into the 10-nl ring with RT/RT-PCR master mix (green);and C:The ring’s peristaltic pump breaks the droplet to mix the cell with master-mix.When the reaction is finished,oil (blue) pushes the product (cDNA) out of the ring in the form of a droplet (10-nL) for downstream molecular analysis (Refer to[17] for details).
MRD monitoring has become one of the most relevant prognostic factors for MM.It has been shown that persistent MRD is associated with improved progression-free survival and overall survival[33].Not only is single cell tracking methodology as described in this article a sensitive method to detect MRD,but the characteristics of the residual cells will also be able to be elucidated.As such,one can detect the emergence of a “dangerous” clone.
Determining the sequencing of therapies including immunotherapy
Currently,we have more than 14 unique treatments for the MM,which,when used in combination,yields dozens of combination options for patients.Clinical trials using antibody drug-conjugate and bispecific antigen-directed CD3 T-cell engager targeting,by checkpoint inhibitors and an anti-T-cell immunoglobulin and ITIM domains antibody are currently underway and has the potential to further prolong survival times[34].Chimeric antigen receptor T cell therapy targeting B-cell maturation antigen,immunoglobulin kappa chain,SLAM family member 7,or G-protein coupled receptor family C group 5 member D,the activated integrin beta7 is a promising treatment modality which can often give long progression-free survival in heavily treated patients[35,36].Despite this arsenal of treatments,the elusive cure for MM has yet to be found,and the current approaches to the treatment of refractory disease produce progressively short-lived efficacy.Perhaps this is because the sequencing of these treatments is often borne out of trial and error and do not take into consideration the temporal changes and spatial relationships of MM subclones.The development of chemoresistance,leading to shorter progression-free with each subsequent treatment and overall survival,lies in understanding how therapies drive the evolution of subclones.Using sensitive methods for detection and characterization of subclones,we can understand whether therapy induces molecular alterations within myeloma cells or selects for the survival of specific clones over others.Knowing how the treatments we use drive the process of evolution can allow clinicians to choose combinations of therapies that will modulate the development of chemoresistance.
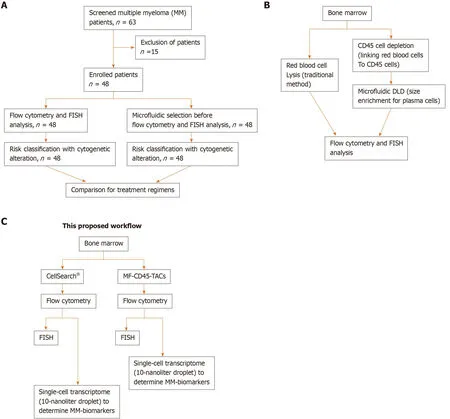
Figure3 Schematic designs of the proposed workflow.A:Consolidated Standards of Reporting Trials diagram.A total of 63 patients were screened for eligibility.Only 48 patients were newly diagnosed with multiple myeloma before receiving any treatment.These patients were enrolled,and their bone marrow obtained at diagnosis was divided into two aliquots:One aliquot underwent traditional flow cytometry and FISH analysis,and the other aliquot was subjected to microfluidic selection for enrichment of CD45-PCs,then subjected to flow cytometry and FISH analysis.Results from both methods were compared;B:Comparison of traditional method to microfluidic method (MF-CD45-TACs).MF-CD45-TACs significantly enrich plasma cells for flow cytometry and FISH assays and improve the accuracy of these assays;C:This proposed workflow (Note that we can use both bone marrow and circulating multiple myeloma cells[76]).
CONCLUSION
We envision that single-cell technology will innovate cancer stem cell subclonal evolution on time-space landscaping of heterogeneity and imply the lineage-tracking pathway-based prediction of therapeutic efficacies of cancer treatment.Accurately,temporal development and spatial distribution of quantitative subclonal measurement of MM will reveal therapeutic sensitizing mutations,thereby moving closer to developing a therapeutic window[37]of cancer in the advent of new,more productive,and less toxic therapies.We hypothesize that subclonal evolution,in conjunction with current standard care,will improve outcomes in patients with heterogeneous pathologies (Figure6)[38].Circulating MM counts and Cav-1 molecules early during radiotherapy are independently predictive of recurrence in MM.Physicians assert that every time that there is a reference to,visual or spoken,the patient view of the landscape of an MM diagnostics that they claim to predict the outcomes legitimately,it has to be as comprehensive and individualized as possible given the data package generated from AI-Med algorithms.Recently,we applied our microfluidic devices to myeloma risk stratification.However,like many current microfluidic devices,the device only enhanced and improved traditional FACS and FISH.The gap of integrating genomic profiles into cancer characterization is still not filled.Therefore,it is a logical and necessary step for us to integrate our single-cell RNA-seq technology into cancer characterization,specifically molecular classification of MM in cancer genome landscape such as the Pan-Cancer Analysis of Whole Genomes consortium[39]."Timing analyses suggest that driver mutations often precede diagnosis by many years,if not decades.Together,these results determine the evolutionary trajectories of cancer and highlight opportunities for early cancer detection"[40].All of these must rely on "A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns"[41]for integration of dynamic space-time changes.One such integrated platform was to scaffold the diverse datasets together,allowing them to interface not only across single-cell transcriptomics (scRNA-seq),but also across distinct cellular modalities –e.g.,a bone marrow atlas to characterize lymphocyte populations[42]– to better understand cellular identity and function beyond the taxonomic listing of clusters of cellular heterogeneity[43].
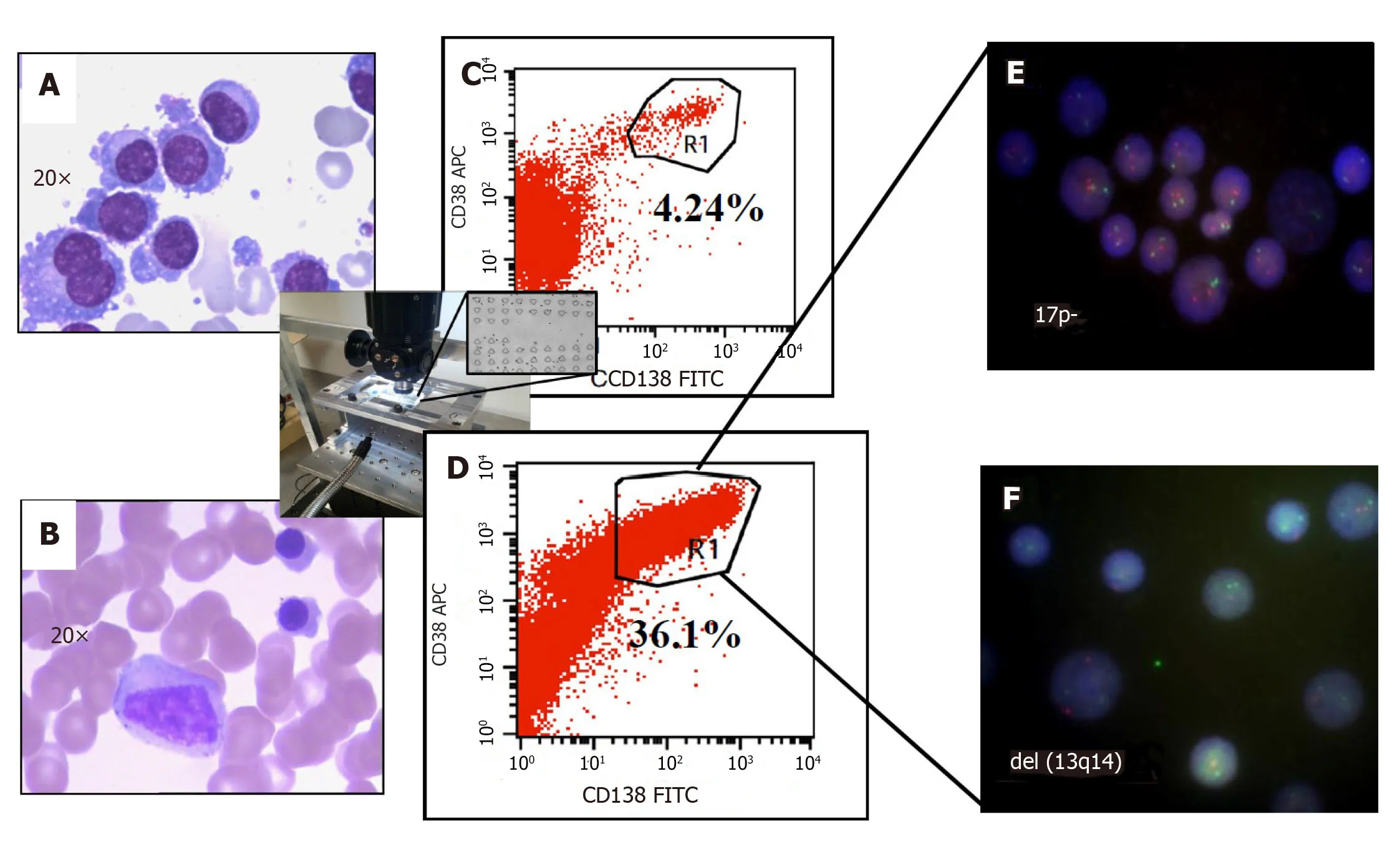
Figure4 Improved clinical outcomes with microfluidic CD45 depletion (Patient 1).A:Bone marrow smear at the time of initial diagnosis;B:Bone marrow smear after effective treatment (complete remission);C:Flow-cytometry without microfluidic enrichment.Plasma cells (CD38+/CD138+) is only 4.24% and no FISH cytogenetic abnormalities were found (low risk);D:After microfluidic enrichment (center inset) plasma cells (CD38+/CD138+) increased to 36.1%;E:FISH on enriched plasma cells show 17p- (Red:D13S319;Green:P53);F:FISH showed del(13q14) in enriched PC (Red:D13S319;Green:RB1).With enriched plasma cell for FISH,the patient was reclassified and treated as high-risk multiple myeloma which leads to complete remission (Refer to[17] for details).
Implementation of this modified diagnostic device in clinics proven to improve clinical outcomes of MM (Figure3C).Our microfluidic-assisted stratification of single cancer cells may help understand the mechanisms underlying the temporal and spatial heterogeneities in solid tumors like brain cancer as testing is underway[44-46],thereby holding promise for using the single-cell analysis to guide treatment for targeted therapy (Table2),as governed by artificial intelligence-based integration of genome,epigenome,and pathological measurements.

Table2 Therapeutics targets and corresponding agents in multiple myeloma and artificial intelligence medicine
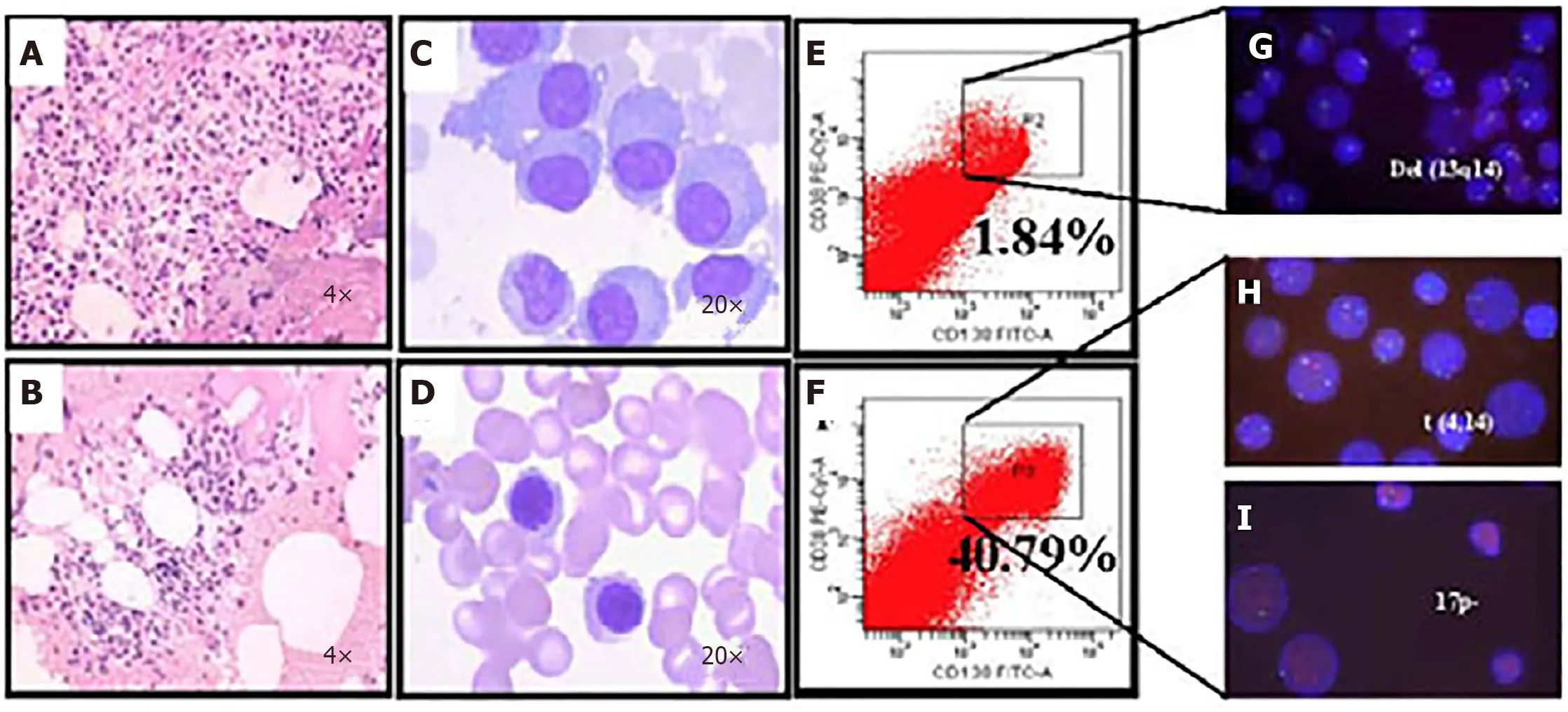
Figure5 Microfluidic risk-stratification improves clinical outcomes of multiple myeloma (Patient 2).A:Bone marrow at diagnosis.Active granulocyte hyperplasia;B:Partial remission was achieved with revised risk-stratification;C:At diagnosis,bone marrow plasma cell (PC) abnormalities included clustered and scattered distribution of primitive and immature PCs,with large cell body,fine chromatin,visible nucleolus,and abundant cytoplasm;D:After treatment for high-risk multiple myeloma,PCs were rare and had normal morphology;no typical abnormal PCs were observed;E:At diagnosis without microfluidic enrichment,PCs (CD38+/CD138+) were only 1.84 %;F:After microfluidic enrichment,PCs (CD38+/CD138+) increased to 40.79%;G:Without microfluidic enrichment,FISH showed IgH rearrangement and del(13q14),leading to classification as intermediate risk (Red:D13S319;Green:P53);H and I:After microfluidic enrichment,in addition to del(13q14),FISH showed t (4,14) fusion (yellow dots) and 17p- (Red:D13S319;Green:P53),patient was reclassified and treated as high-risk,which led to efficacious treatment (Refer to[17] for details).
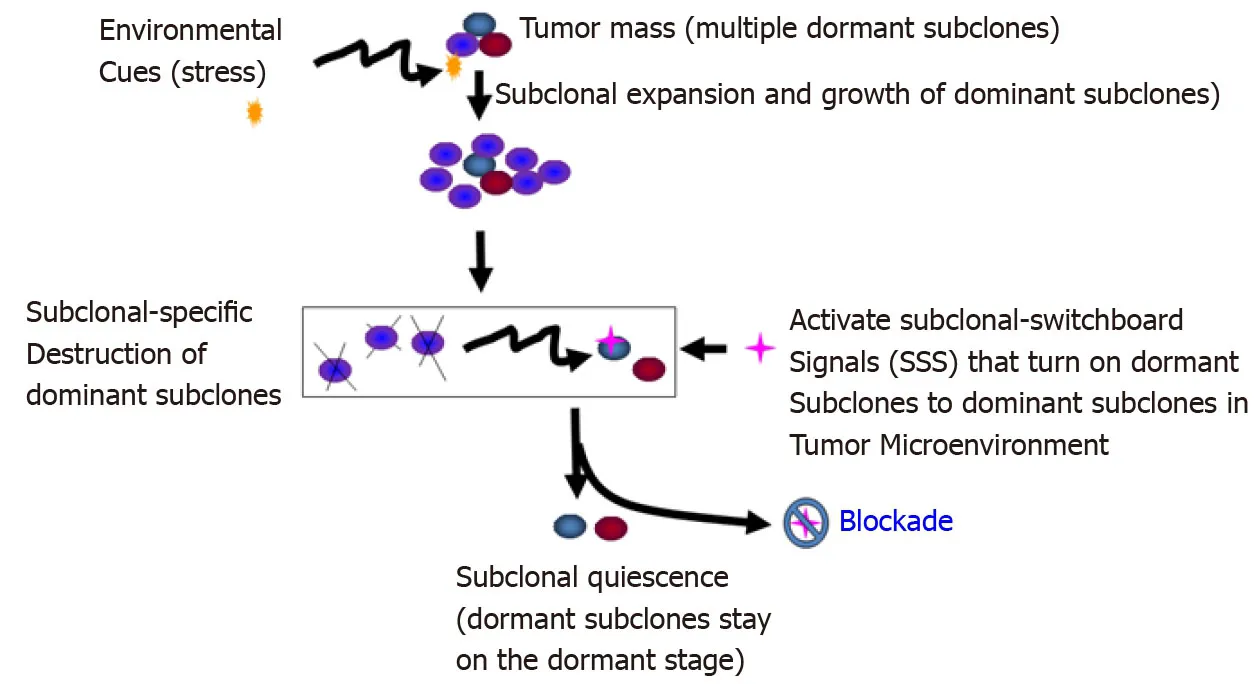
Figure6 Blockade of the dominating subclonal switchboard signals in cancer stem cells as a new therapeutic strategy to suppress the dominating subclone shift to control cancer progression and post-treatment cancer recurrence.Showed is the proposed new treatment paradigm that should target the subclonal-switchboard signals (SSS).Blocking the dominating subclonal SSS leads to subclonal quiescence,so keeping tumors alive but small and manageable (dormant/quiescent subclone).Note that SSS as mechanisms for leading to shifting dominating subclones as triggered by environmental cues(stress) for cancer progression and post-treatment.A cancer subclone may gain a mutation that,in the appropriate environment cue,leads to dominating subclonal activation due to positive selection.Showed lettering and lines/ arrows in the black color is the current concept of a treatment strategy for cancer- dominant subclonal cells (cancer stem cells) that may acquire a mutation in a suitable environment,triggering to dominating subclonal expansion and growth.When this dominating subclone is explicitly destroyed,it sends out dominating subclonal-SSS to a dormant/quiescent subclonal cell,which gets activated for dominating subclonal expansion and growth (adopted from[38]).
ACKNOWLEDGEMENTS
We thank Maria Minon,MD;Brent A Dethlefs;Mustafa H Kabeer,MD;William G Loudon,MD,PhD;Leonard S Sender,MD;Anthony Christopher Chang,MD,MBA,MPH;Edward Nelson,MD;Richard A van Etten,MD,PhD;Dan Cooper,MD;and Jiang F Zhong,PhD;for their support and enthusiasm.
 World Journal of Stem Cells2020年8期
World Journal of Stem Cells2020年8期
- World Journal of Stem Cells的其它文章
- Role of mesenchymal stem cell derived extracellular vesicles in autoimmunity:A systematic review
- Human embryonic stem cell-derived mesenchymal stem cells improved premature ovarian failure
- Assessment of tobacco heating system 2.4 on osteogenic differentiation of mesenchymal stem cells and primary human osteoblasts compared to conventional cigarettes
- Mesenchymal stem cell-derived exosomes:Toward cell-free therapeutic strategies in regenerative medicine
- Autophagy in fate determination of mesenchymal stem cells and bone remodeling
- Human embryonic stem cells as an in vitro model for studying developmental origins of type 2 diabetes