
线粒体非典型功能障碍与神经系统疾病相关研究进展*
2020-09-16 03:12:34杨皓翔综述孔庆霞审校
济宁医学院学报 2020年4期
杨皓翔 综述 孔庆霞 审校
(济宁医学院临床学院,济宁 272013;济宁医学院附属医院,济宁 272029)
线粒体主要功能是为细胞提供三磷酸腺苷(ATP),此外还有调节细胞凋亡、产生氧化剂、维持钙平衡等非典型功能。线粒体依靠完整的呼吸链氧化磷酸化过程为机体供能,该过程相关蛋白由线粒体基因与核基因共同编码[1]。大脑占机体静息状态下耗氧量的20%,更加依赖线粒体的功能完整性,而线粒体功能障碍与神经系统疾病有着复杂的关系。近年来随着研究的深入,发现线粒体非典型功能对神经系统疾病的发生、发展也有影响。神经系统相关致病基因会破坏线粒体的形态、结构和功能,线粒体功能受损又进一步造成神经系统损害,二者之间恶性循环。对线粒体非典型功能机制的探究有助于理解线粒体疾病,对线粒体功能障碍干预治疗,是神经系统疾病临床诊治的新思路。
1 线粒体非典型功能及生理机制
线粒体在脑组织与肌肉组织含量最为丰富,除ATP合成以外,其它非典型功能也参与机体正常的理化调节,维持理化环境稳。见图1。
1.1 调节细胞凋亡
细胞凋亡是程序性死亡,线粒体功能障碍与其密切相关[2]。线粒体通过细胞色素C(Cyt-C)、凋亡诱导因子(apoptosis induced factor,AIF)、BCL-2、Caspase家族等蛋白介导细胞凋亡。线粒体损伤后,外膜蛋白聚集,Cyt-C通过线粒体膜通透性转换孔(mitochondrial permeablity transition pore,MPTp)进入胞质,依赖Caspase途径来诱导细胞凋亡[3]。AIF是一种黄素蛋白,引起线粒体肿胀,改变外模通透性,AIF和Caspase-12进入细胞核后诱导DNA片段化,促使细胞凋亡。但目前针对AIF是否依赖Caspase途径仍有争议,未来需要进一步明确具体通路。生理情况下BCL-2及其亚型通过抑制线粒体内膜超级化来维持外模完整,并与阴离子通道结合阻止钙离子进入胞质,抑制MPTp释放Cyt-C[4]。Caspase家族是一类特异性蛋白酶,Caspase-3位于通路下游,是通路的中心环节。靶向线粒体KillerRed(mtKR)通过Cyt-C/Caspase-3途径促进活性氧(reactive oxygen species,ROS)产生,Cyt-C从线粒体进入胞质激活Caspase-3/Caspase-9导致线粒体功能障碍,引起细胞凋亡[5]。因此,未来研究神经系统退行性病变,可从线粒体调节细胞凋亡层面寻找临床治疗靶点。
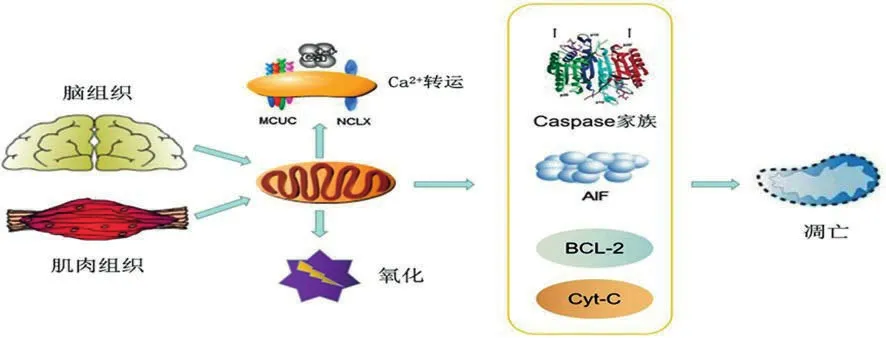
注:脑组织与肌肉组织生理活动高度依赖线粒体,线粒体非典型功能包括调节凋亡、稳定钙离子转运、产生氧化剂等,通过Cyt-C、BCL-2、AIF、Caspase家族等调节细胞凋亡;通过MCUC-NCLX联合稳定钙离子,维持钠-钾化学浓度梯度;通过呼吸链反向转移电子产生氧化剂等。
1.2 产生自由基和氧化剂
线粒体产生氧化剂在细胞信号传递过程有重要作用,积极参与细胞内的生理活动。在线粒体中发生大量的电子转移反应,若一个电子转移至氧则产生自由基、超氧自由基[6],进而产生过氧化氢、羟基自由基、过氧亚硝基等氧化剂。线粒体氧化剂的产生具有组织特异性,在大脑中主要是反向电子转移,电子从辅酶Q转移至复合体I,把氧还原成超氧自由基,这个过程受钙离子调节[7-8]。Tahara等[8]报道,线粒体内膜电位调控氧化剂的产生,高膜电位促进氧化剂产生,低膜电位则抑制氧化剂产生。在高内膜电位条件下,大脑线粒体产生氧化剂过程与Cyt-C释放有关,Cyt-C损害了电子流动,促进了超氧自由基的形成。线粒体自由基产生是偶然发生的,自由基的产生伴随生命进化而存在,所以线粒体产生氧化剂也参与了细胞内的生理反应,其功能紊乱与神经系统疾病发生有关。因此,线粒体产生自由基并不总是有害的,自由基能影响Cyt-C释放,对于钙离子稳态有反馈作用。
1.3 调控钙离子缓冲
线粒体钙离子转运障碍与神经系统疾病有关。线粒体钙统一转运蛋白复合体(MCUC)附着在内膜上,借助内膜驱动力把钙离子从胞浆转运到基质[9]。MCUC门控是由MICU-2和MICU-3介导,MICU使突触前膜线粒体对Ca2+震荡更敏感,这是神经元在激活状态下保证ATP供能的关键[10]。线粒体在钠钙锂泵(NCLX)介导下排出钙离子,吸入钠、氢离子,维持钠-氢化学浓度梯度,线粒体等细胞器对胞质内超载钙离子的摄取,在细胞钙稳态的生理和病理变化中起到重要作用[11],内质网-线粒体接触点破坏后会使钙离子稳态丧失,增加细胞死亡的易感性[12]。钙离子缓冲还受到线粒体形态和动力学的调节,人工高灌流线粒体具有更强的钙摄取能力,线粒体分裂则会使摄取速度和摄取能力极大下降[11]。正己烷引起线粒体融合-分裂失衡,骨骼肌线粒体稳态向分裂转移、代谢紊乱,导致周围神经和骨骼肌损伤[13]。因此,完整的线粒体形态和其它细胞器动力学运输作用,在神经元生理活动中至关重要,具体机制仍有待进一步研究。
2 线粒体非典型功能障碍与神经系统疾病
2.1 缺血性脑卒中
缺血性脑卒中是死亡和致残的主要原因之一。细胞缺血缺氧造成复合体I抑制、琥珀酸盐积累、线粒体生成大量超氧自由基,引起脑组织损伤。缺血缺氧时巯基残基氧化使酶失活,复合体I受抑制,其功能受损[12]。同时,线粒体琥珀酸积累,导致琥珀酸脱氢酶逆转,在此条件下由富马酸形成琥珀酸,同时电子反向转移至复合体I,产生超氧自由基,进而生成大量氧化剂,因此,防止琥珀酸积累、使用线粒体靶向抗氧化剂都可以显著保护脑组织损害。线粒体DNA水平在脑损伤后几小时内表达升高[14],这表明线粒体可能参与缺血性脑卒中损伤修复。线粒体是氧化剂损害的靶细胞器,受损细胞通过星形胶质细胞、间充质干细胞的线粒体来重新填充自己受损的线粒体[15],但供体和受体之间如何转运线粒体等细胞器,其机制仍有待进一步探究。保持线粒体形态完整对神经功能具有保护作用,更多的线粒体融合具有钙离子缓冲能力。维持线粒体功能和形态完整,对预防缺血性脑卒中后脑组织损伤以及卒中后修复有重要意义。
2.2 亨廷顿舞蹈症
亨廷顿舞蹈症患者线粒体电子传递活性下降、钙离子摄取能力受损、铁稳态改变、氧化剂产生、自身形态及调节改变。亨廷顿蛋白基因编码区CAG序列重复扩增,造成神经元丢失,该病的神经元损伤与线粒体功能异常有关[15-16]。线粒体维持生理能量代谢需要亨廷顿蛋白参与。呼吸链受抑制复合体II活性降低,导致线粒体膜电势降低。在亨廷顿动物模型中3-硝基丙酸(3-NPA)抑制呼吸链复合物II[16],引起纹状体细胞毒性,进一步影响线粒体生理活动,这表明呼吸链复合物活性降低是亨廷顿舞蹈症线粒体功能障碍的原因之一。在脑组织中线粒体钙摄取能力受损,铁稳态改变引起亨廷顿舞蹈症病理变化[17],CAG序列大量扩增会导致线粒体对钙离子摄取降低,钙离子摄取能力强弱与线粒体膜亨廷顿蛋白含量直接相关。纹状体线粒体摄取钙离子能力越强,越能对神经元起到代偿性保护作用。纹状体神经元和星形胶质细胞与皮层部位不同,纹状体神经元钙离子缓冲能力一般较弱,对钙离子稳态失衡更加敏感。Joshi等[18]报道,线粒体氧化失衡、形态和周转变化是亨廷顿舞蹈症的重要原因之一。通常来讲,线粒体功能异常引起亨廷顿舞蹈症的同时也伴随纹状体损伤,这可能是纹状体星形胶质细胞特殊性导致,纹状体细胞代谢氧化大量的脂质,产生更多的氧化剂。目前仍有许多问题亟待解决,例如:改良的亨廷顿蛋白促进神经退化的确切机制是什么?为什么这种疾病和纹状体对复合体II的抑制特别敏感?总体而言,针对线粒体呼吸抑制治疗是临床上亨廷顿舞蹈症一个新的、有前景的治疗靶点。
2.3 肌萎缩侧索硬化症(amyotrophic lateral slerosis,ALS)
ALS是运动神经元进行性变性病。ALS相关致病基因突变后,影响线粒体等细胞器的生理功能,如细胞骨架动力学、RNA的转运、蛋白质稳态等,进而引起内质网应激、钙离子信号中断、蛋白聚集、轴突运输障碍、线粒体功能障碍和细胞凋亡等。ALS患者中90%为散发型,10%为家族型,基因C9ORF72、SOD1、FUS、CHCHD10等突变与ALS有关(见表1)。散发型ALS患者在运动神经元中发现异常线粒体,线粒体功能障碍导致复合体IV活性受损[19],散发型和家族型ALS患者中均出现成纤维细胞中线粒体广泛改变,例如:线粒体膜电位、耗氧率、呼吸链复合体的活性和水平、ATP、活性氧和钙离子水平等[20]。此外,ALS致病基因突变对线粒体功能影响不尽相同。基因R495X突变后RNA翻译中断,并表达较小的线粒体突起,R495X突变后与成熟的mRNA结合,野生型则与前体mRNA结合[21]。基因CHCHD10编码线粒体蛋白,作用于线粒体接触部位的嵴连接处和嵴组织系统,还参与铁代谢过程[22]。基因CHCHD10发生获得性突变后,线粒体结构变化,进而导致线粒体能量代谢障碍[22]。ALS患者的成纤维细胞中线粒体高灌注并且依赖无氧糖酵解[23],在此条件下复合体I会受到糖酵解的损害。综上所述,导致ALS的因素众多,目前确切机制尚不完全清楚,仍需进一步深入研究线粒体功能障碍与ALS发生关系。

表1 肌萎缩侧索硬化症相关致病基因突变
2.4 帕金森病(Parkinson’s disease,PD)
PD主要是黑质多巴胺能神经元进行性丧失,导致运动症状和非运动症状。研究表明,线粒体异常是神经元丢失的触发因素,进而产生这种疾病的临床症状[24]。1-甲基-4-苯基-1,2,3,6-四氢吡啶(MPTP)及其代谢产物会抑制复合体I,复合体I缺乏导致黑质神经元丢失引起帕金森病,造成线粒体钙离子稳态改变和氧化失衡[25]。PD的致病基因突变也会引起线粒体功能障碍。Parkin蛋白是一种E3泛素连接酶,它泛化线粒体膜外膜蛋白,生理情况下促进受损线粒体清除,基因Parkin突变导致线粒体清除障碍[26]。PTEN诱导激酶1(PINK1)突变后导致常染色体隐性遗传帕金森,该基因编码线粒体中丝氨酸激酶,与野生型相比,基因敲除组的基础呼吸率显著降低,多巴胺释放减少[27],因此纹状体中多巴胺释放减少可能由线粒体功能障碍所致。Kostic等[28]报道,敲除基因PINK1会抑制线粒体钠钙交换蛋白(NCLX)的活性。蛋白激酶LRRK2由基因PARK8编码,LRRK2损伤与帕金森有关,也影响NCLX的功能[29],这表明钙离子外流可能是帕金森病理变化的潜在原因。因此,线粒体功能障碍与帕金森病病理生理机制、致病基因突变等相关,这可以为帕金森治疗提供新的靶点。
3 小结与展望
目前对线粒体功能障碍与神经系统疾病相关研究已取得较大进展,但绝大多数研究局限于线粒体产生ATP层面,线粒体的其它功能也在神经系统疾病发生、发展过程中发挥至关重要的作用。缺血性脑卒中患者,脑细胞中氧与底物灌注减少、代谢物的积累,最终导致线粒体损伤和细胞死亡。振动病动物模型发现,骨骼肌缺血缺氧时线粒体呼吸链酶破坏,引起钠-钾泵、钙泵异常,造成周围神经系统和骨骼肌损伤[30]。神经系统退行性病变与线粒体功能障碍关系更为复杂,二者因果关系尚不确切,无论线粒体作为神经系统退行性病变的诱因,还是直接参与神经元损伤,对线粒体的保护性干预都是潜在治疗靶点。在分子生物学层面,线粒体基因与核基因突变均会导致线粒体病,线粒体途径是如何诱导神经元损害,仍需要进一步完善基础实验明确机制。在已知基因和通路的基础上,探究线粒体与神经系统之间的复杂关系,进一步发现新的基因与通路,深入剖析促进-抑制调控网络,寻找新的靶点,为临床提供更精准的靶向治疗。
猜你喜欢
中学化学(2019年4期)2019-08-06 13:59:37
中学化学(2019年4期)2019-08-06 13:59:37
科学24小时(2019年4期)2019-06-10 10:17:53
浙江农业学报(2017年1期)2017-05-17 06:13:45
科学生活(2016年7期)2016-07-25 12:41:35
原子与分子物理学报(2015年1期)2015-11-24 12:49:26
河南医学研究(2014年4期)2014-02-27 14:52:25
食品科学(2013年15期)2013-03-11 18:25:51
地球学报(2012年1期)2012-09-20 00:46:42
中国医学科学院学报(2012年3期)2012-03-25 13:58:59
