
Porous Bioactive Ceramic Derived from Polyurethane Foams
2020-09-15 11:46ZHANGYuanyuanLUOMinhuaXIAOZhuohaoZHUQingxiaLIUSaliCHENHuilingGUORulian
陶瓷学报 2020年4期
ZHANG Yuanyuan , LUO Minhua , XIAO Zhuohao , ZHU Qingxia , LIU Sali ,CHEN Huiling , GUO Rulian
(1. Jingdezhen Ceramic Institute, Jingdezhen 333403, Jiangxi, China; 2.Tiangong University, Tianjin 300380, Tianjin, China)
Abstract: Bioceramic matrix powders were synthesized by using a sol-gel method, while water based slurries were prepared by adding lab-made flux, polyvinyl alcohol (PVA), carboxyethyl cellulose and n-butanol. Polyurethane foams after hydrolysis and surface modification were impregnated in the slurries and the excess slurries were removed. The samples were then dried and sintered at high temperatures to obtain bioactive ceramics. The effects of PVA content, self-prepared flux content and sintering temperature on porosity and flexural strength of the samples were systematically studied. The pore samples were analyzed by using scanning electron microscope (SEM), their biological activity was examined by using simulated body fluid (SBF), and the phase changes before and after mineralization were characterized by using X-ray diffraction (XRD). It was found that porous bioactive ceramics with promising comprehensive properties can be obtained by adding 30 wt.% flux and 0.76 wt.% PVA, with a sintering temperature of 1000 °C. The porosity of the optimized sample is 81.35% and the flexural strength is 2.75 MPa.
Key words: porous bioceramic; organic foam impregnation method; porosity; biological activity; flexural strength
1 Introduction
Bioactive ceramics prepared by using sol-gel methods are of special compositions, high bioactivity and biocompatibility, thus having wide applications in bone tissue reconstruction[1]. Since biological scaffolds made of bioactive ceramics have large porosities with interconnected pores, the smaller pores are responsible for transport of nutrition and discharge of wastes, while the larger pores provide channels and space for the growth of osteoblasts,when they are implanted into human bodies, thus ensuring the rapid growth of blood vessels and tissue cells and promote the development of bone tissues[2]. There are various ways to fabricate bioactive scaffolds. The use of pore agents is a simple method to obtain bioactive scaffolds, but the pore sizes are not uniform, so that it is difficult to form 3D interconnected channels and they have relatively low mechanical strengths[3,4]. Foaming methods have disadvantages of uncontrollable pore structures, poor pore size distribution and low pore connectivity[5-7].Freeze drying methods result in directionally controllable pores, but the pore connectivity and strength are relatively low, while the pore sizes are small[8,9]. 3D printing technologies require slurries with high contents of liquid phases, which cause deformation of green bodies due to the effect of gravitation. Meanwhile, the samples are prone to crack and deform during binder burning and sintering[10,11]. In comparison, foam conversion process is of various advantages, such as simple operation, cost-effectiveness, high porosity and high pore connectivity, thus attracting strong attentions all around the world[2,12-14]. However, there are still rooms for further improvement. For instance, as the porosity is higher than 80%, compressive strengths of the scaffolds will be only in the range of 0.3-0.4 MPa[15-17]. Baino et al.[18]introduced organic components to increase the strength of scaffolds, but the highest compressive strength as still only 1.0 MPa, below the requirement of bone regeneration.Although the mechanical strength could be increased by increasing the sintering temperature,this process would also reduce the bioactivity of the scaffolds. Therefore, it is still a challenge to fabricate bioactive ceramic scaffolds with sufficiently high mechanical strengths, while not compromising their porosity and bioactivity[19].
In this study, a sol-gel process was employed to prepare bioactive ceramic scaffold through foam conversion, which exhibited sufficiently high porosity, strong mechanical properties and relatively high bioactivity, by adjusting the contents of PVA and lab-made flux.
2 Experimental
2.1 Materials
In the sol-gel process, chemicals and reagents include anhydrous ethanol (C2H6O, AR), tetraethyl oxysilicate (C8H20O4Si, AR), triethyl phosphate(C6H15O4P, CP), citric acid (C6H15O4P, CP), calcium nitrate tetrahydrate (Ca(NO3)·4H2O, AR) and polyethylene glycol 400 (H(OCH2CH2)nOH, CP).Chemicals for bioactive ceramics include polyvinyl alcohol ([-CH2CHOH-]n/(C2H4O)n, AR), carboxyethyl cellulose (C2H6O2·X, CP) and n-butanol (C4H10O,AR). Properties of the polyurethane foams used in the experiment are listed in Tab. 1. Sodium hydroxide (NaOH, AR), tween 80 (C24H44O6, CP)were used to pretreat the polyurethane foams.
The chemical reagents used to prepare SBF include sodium chloride (NaCl, AR), sodium bicarbonate (NaHCO3, AR), potassium chloride(KCl, AR), dipotassium hydrogen phosphate (K2HPO4·3H2O, AR), magnesium chloride hexahydrate(MgCl2·6H2O, AR), hydrogen chloride (HCl, AR),calcium chloride (CaCl2, AR), sodium sulfate(Na2SO4, AR), and trihydroxymethylaminomethane((CH2OH)3CNH2, AR).
2.2 Sol-gel process to prepare bioactive ceramics
320 mL deionized water and 50 mL anhydrous ethanol were mixed at 30 °C, while 1.5 g citric acid was dissolved into the mixture with the aid of stirring. 40 mL TEOS and 3.5 mL triethyl phosphate were mixed, according to the composition of the bioceramics, i.e., 60% SiO2, 4% P2O5and 36% CaO. In the mixture, 31 g calcium nitrate tetrahydrate was slowly added. Then, 2 mL polyethylene glycol 400 was dropped to foam sol.The sol was sealed and kept in a water bath then aged 24 h into gel, dried at 48 h for 70 °C. The dried gel was calcined at 600 °C for 2 h with a heating rate of 5 °C /min in a muffle furnace, and thenbioactive ceramics were obtained.
2.3 Selection and pretreatment of polyurethane foams
Organic precursor is critical to the fabrication of porous scaffolds. The selected polyurethane foam is of 2D interconnected porous structure, strong resilience, wide volatilization temperature range(200-600 °C), high temperature volatilization temperature (380 °C) and low softening temperature(250 °C)[20]. As a result, it is softened gradually during the thermal decomposition process, thus preventing the samples from deformation during the volatilization of the organic components. In order to avoid the occurrence of plugging in the porous scaffolds due to excessive inter-membranesin the polyurethane foam, also owing to reduced slurry adhesion on the smooth surface of the polyurethane foam, it should be pretreated with alkaline solutions[21]. In this study, water was used as the solvent; the wettability of the polyurethane foam by the slurry after the alkaline treatment was still insufficient, thus leading to too thin and too thick coatings of slurry at the cross section and corner line,respectively[22]. This occurrence would result in serious cracking of the scaffolds, thus deteriorating their mechanical strengths. Surfactant is a hydrophilic substance, which can be used to improve the hydrophilicity of polyurethane foam and hence reduce the surface/interface tension[23]. As a consequence,water-based slurries can be firmly adsorbed onto the polyurethane foam, so as to improve the wettability between polyurethane foam and slurry.
To measure flexural strengths, the polyurethane foam was cut into strips with sizes of 40 mm × 15 mm × 15 mm. The strip samples were immersed in 0.6 mol/L NaOH solution for 2 h and then washed with distilled water to neutral pH, followed by drying. The dried samples were kept in tween 80 solution for 2 h and dried again.
2.4 Preparation of porous bioactive ceramics
52.5 g bioactive ceramic powder and lab-made flux were ball milled separately and sieved through 250 mesh. The milled samples were mixed with 100 ml distilled water, followed by adding 0.5 g carboxyethyl cellulose and stirring for 30 min. Then,PVA solution (PVA was dissolved in 100 mL distilled water at 90 °C with constant stirring) and stirred for 1 h, followed by dropping 1 mL n-butanol and stirring for 24 h to foam slurry. Pretreated polyurethane foam strips were entirely soaked in the slurry. The soaked samples were taken out and squeezed to remove the excessive slurry, followed by drying at 30 °C for 12 h and 60 °C for 12 h at a heating rate of 0.5 °C /min. The dried samples were sintered at 1000 °C, 1100 °C and 1200 °C for 2 h, at heating rate 2 °C /min. The contents of lab-made flux and PVA are listed in Tab. 2.

Tab.1 Properties of the polyurethane foam
2.5 Preparation of SBF and bioactivity characterization
Bioactive materials are defined as those that have chemical bonding with living bone tissues and soft tissues[24,25]. The chemical bonding is formed through the deposition of hydroxycarbonate apatite(HCA) at interfaces between the biomaterials and HCA, because HCA is similar to the basic mineral hydroxyapatite (HA) in structure and properties. In addition, HCA has relatively high solubility in body flow and hence degradable in human body, thus have high bioactivity[26]. Moreover, biological proteins can be adsorbed onto HCA to form bone cells, thus facilitating bone guidance and ensuring the chemical bonding between the bioactive materials and the bones[25]. The binding capability of biomaterials to host bones is closely related to the presence of the HCA layer formed on surface of the biomaterials. Therefore, the biological activity of biomaterials can be preliminarily evaluated by monitoring the growth of HCA on the surface of biomaterials by immersing them in SBF solution[27].

Tab.2 Slurry formula in terms of the contents of lab-made flux and PVA
Lab-use SBF contained no cell and protein, with a pH value of 7.4. The contents of ions are listed in Tab. 3, which are similar to those of human blood plasma. To prepare the SBF[28], 8.035 g NaCl, 0.355 g NaHCO3, 0.255 g KCl, 0.231 g K2HPO4·3H2O,0.311 g MgCl2·6H2O, 0.292 g CaCl2and 0.072 g Na2SO4were successively put in 900 mL distilled water, with constant stirring at 37 °C. The solution was adjusted to pH value of 2.0 ± 1.0 with 1 mol/L HCl solution, followed by adding 6.118 g(CH2OH)3CNH2with stirring. Finally, pH value was adjusted to 7.4 and the solution was transferred to a 1000 mL flask.

Tab.3 Ion concentration in human plasma and the SBF solution (mmol /L)
The porous samples were immersed in SBF solution in polyethylene bottles, according to the ratio of sample mass to SBF solution volume of 1 mg/mL. The bottles were then sealed and statically kept at 37 °C, while the SBF solution was replaced every two days. After reaction for different times (2,5, 10 days), the samples were taken out and washed with acetone for one time and deionized water for 3-4 times, followed by drying at 40 °C and sterilization at 120 °C.
2.6 Characterization
Porosity of the samples was measured by using a volume-mas tester. Phase composition was examined by using XRD (D8 Advance, Bruker AXS,Germany), at voltage of 40 kV and current of 40 mA,with scanning step of 0.2 °. Pore structure and mineralized morphology were characterized by using field emission scanning electron microscopy(JSM-6700F, JEOL). Flexural strength was tested by using a pressure tester (FC056R), with a loading rate of 0.5 mm/min. Viscosity of the slurry was measured by using a digital rotational viscometer(LVDV-1), over 10-600 Pa·s. Crystal size was estimated by using the Scherrer formula[29]:

whereDis crystal size,k= 0.89,λis the wavelength of X-ray,βis full width at half maximum(FWHM) of diffraction peaks andθis the diffraction angle.
3 Results and Discussion
3.1 Slurry viscosity
Fig. 1 shows viscosities of the samples with different contents of PVA (0 wt.%, 0.57 wt.%, 0.76 wt.%, 0.95 wt.%) and flux agent (0 wt.%, 10 wt.%,20 wt.%, 30 wt.%). Obviously, the effect of flux agent on viscosity of the slurries was relatively weak. The viscosity was gradually increased with increasing content of PVA, with the maximum value to be 5.3 Pa·s (with flux of 30 wt.%). The viscosities were increased by 32-37% and 22-25%, as the contents of PVA were increased from 0.57 wt.% to 0.95 wt.% and 0.76 wt.% to 0.95 wt.%, respectively.This is because PVA promoted the connection of the ceramic particles through either chemical or molecular bonding, thus reducing the flowability and hence increasing the viscosity of the slurries.
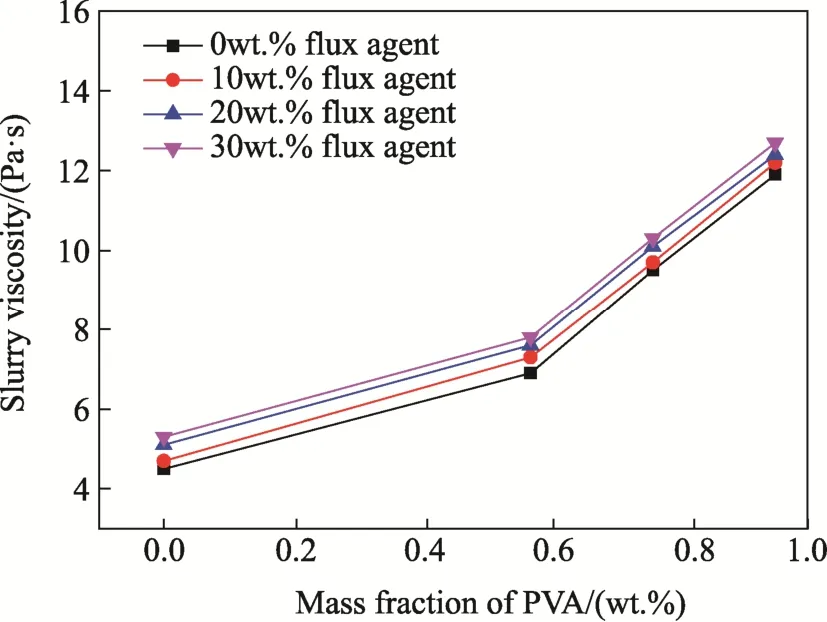
Fig.1 Viscosities of the slurries with different contents of flux as a function of the content of PVA
3.2 Porosity
Fig. 2 shows porosities of the porous samples.For the samples with same content of flus after sintering at same temperature, the viscosity followed the same variation trend, which was increased first and then decreased. As the PVA content was 0.76 wt.%, porosity of the samples was maximized.Initially, with increasing content of PVA, the viscosity of the slurries was increased, so that cohesion of the slurry onto the polyurethane foam was enhanced. As a result, the slurries would not flow into the pores, thus leading to samples with high porosity. However, if the viscosity was too high,the excessive slurries could not be completely removed from the foam, so that the porosity was decreased. With same contents of flux and PVA, the porosity was significantly reduced with increasing sintering temperature, simply because sintering resulted in shrink of the samples.
With fixed content of PVA and sintering temperature, the effect of flux on porosity was not very significant. As the content of PVA was 0.57 wt.% and the sintering temperature was 1200 °C, the highest porosity was 74.19% and the lowest one was 72.24%, with a difference to be only 2.6%. This is simply because the flux had almost no effect on the viscosity of the slurries. Also, the ceramic bodies had been nearly fully densified after sintering at temperatures of ≥1000 °C. At the sintering temperatures, the molten flux would fill the pores.Therefore, porosity of the samples was mainly determined by the porosity of the polyurethane foams.
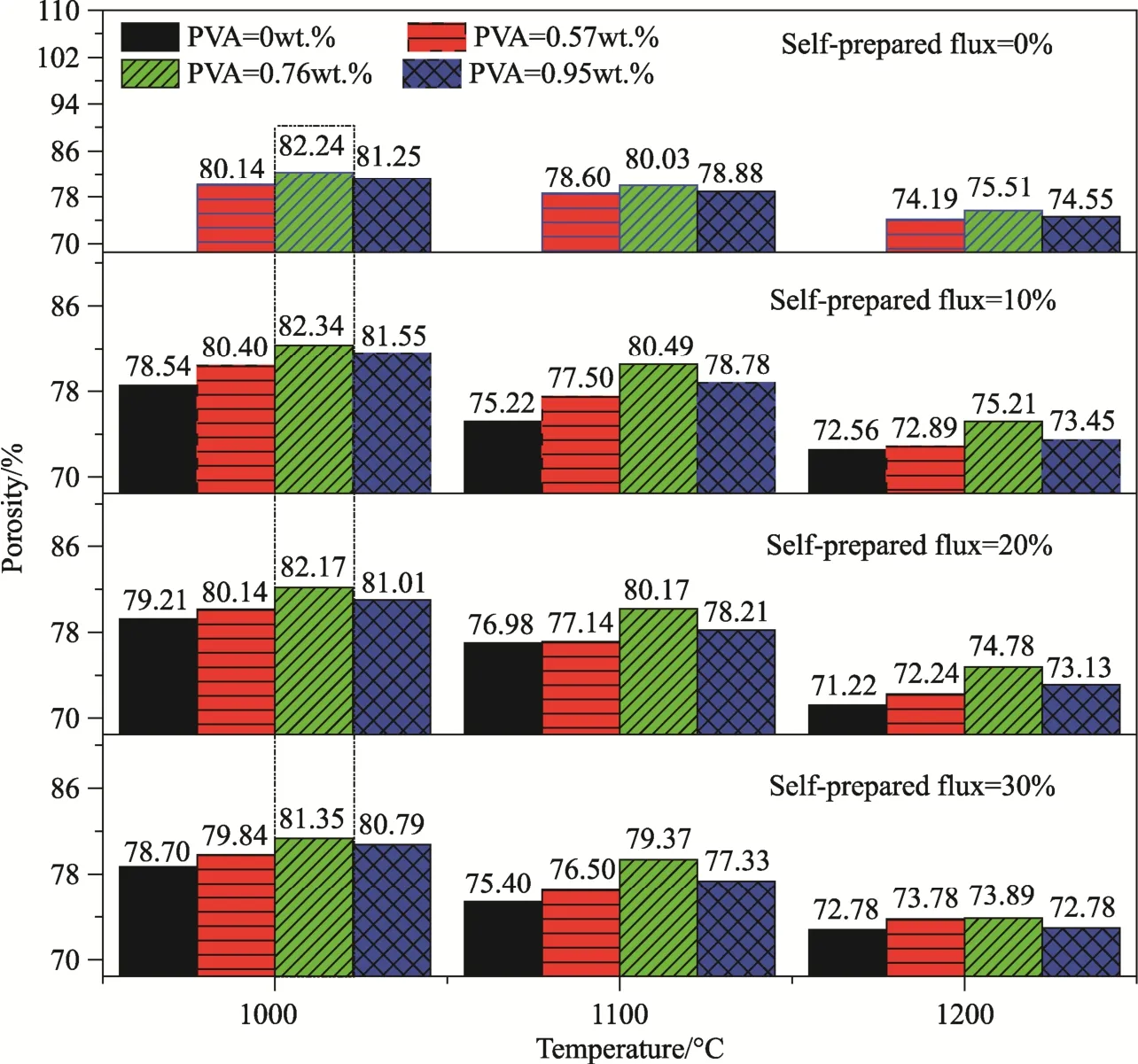
Fig.2 Porosities of the samples with different contents of PVA and flux after sintering at different temperatures
3.3 Flexural strengths
Fig. 3 shows flexural strengths of the porous samples. With given content of flux and sintering temperature, the variation trends in flexural strength were similar over the PVA content range of 0-0.95 wt.%. The increase in flexural strength was quite fast initially and the slowed down. However, without the use of PVA (0 wt.%), the sample had obviously very low strength. This is because the PVA absent slurry had too low viscosity, so that the content adsorbed by the foam was very low. As a consequence, the ceramic layers were too thin and hence the flexural strength was low. With the addition of PVA, the quantity of the slurry adsorbed by the foam was increased, so that the thickness of the ceramic layer was increased. Accordingly, the flexural strength was increased. However, the magnitude of increase in flexural strength as a function of PVA content was almost neglectable. For instance, as the content of PVA was increased from 0.57 wt.% to 0.95 wt.%, the flexural strength was increased by only ≤ 5%. With fixed contents of flux and PVA, the flexural strength was greatly increased with increasing sintering temperature, simply because of the increased density of the samples.
For the samples with same content of PVA after sintering at same temperature, their flexural strength was rapidly increased first and slowly went up later,as the content of flux was increased from 0 wt.% to 30 wt.%. Without the addition of flux, the sample had a very low flexural strength. This is because the ceramic powder could not be fully densified during the sintering process, without the presence of liquid phase (flux). The maximum strength was only 0.73 MPa. The liquid phase was filled in the pores in between the ceramic particles, promoting the densification of the samples. The magnitude of the increase in flexural strength was about 10%.
It is accepted that the minimum compressive strength required for cancellous bone is 2 MPa[30],because flexural strength of ceramics is usually lower than their compressive strength. As seen in Fig. 3, except the ones without the addition of PVA and flux, all the samples meet the minimum strength requirement. Close inspection of Fig. 2 revealed that the four samples with 0.76 wt.% PVA after sintering at 1000 °C exhibited higher porosities (data in the dotted line box) than other ones. Taking into account the porosity and flexural strength, the sample with the highest porosity of 82.34% had a flexural strength of 2.34 MPa, while the sample with highest flexural strength of 2.75 MPa possessed a porosity of 81.35%. In other words, the flexural strength was increased by 17.5%, while the porosity was only reduced by 1.2%. In this case, the content of flux was 30 wt.%. Because low sintering temperature is beneficial to bioactivity[31], the sample with 0.76 wt.% PVA and 30 wt.% flux after sintering at 1000 °C (12# in Tab. 2) was used for the characterization of pore structure and mineralization.
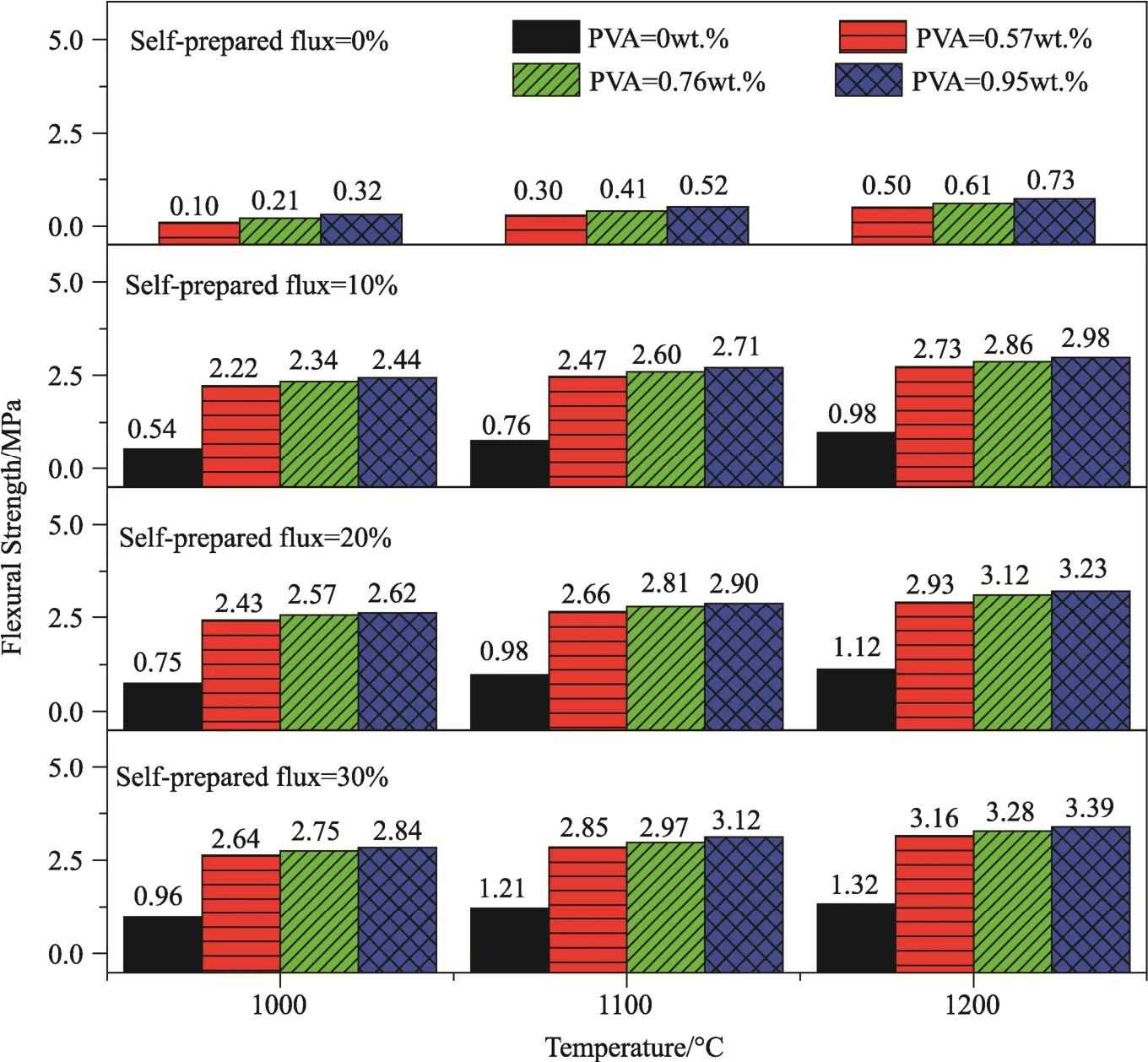
Fig.3 Flexural strengths of the samples with different contents of PVA and flux after sintering at different temperatures
3.4 Pore structure and size distribution
Fig. 4 shows SEM images of the sample 12#. As illustrated in Fig. 4(a), the sample contained a large number of pores with sizes at millimeter scale, which were attributed to the porous structure of the polyurethane foam. The pores were entirely interconnected, with pore sized to be in the range of 100-700 μm at the connecting paths. The sizes of pores suitable for the growth of non-mineral bone tissues, bone tissues, new bone and blood vessels are 40-100 μm, 150 μm, 200-400 μm and ≥500 μm,respectively[32,33]. Meanwhile, connectivity of the scaffold structure is also of crucial effect on the growth of tissues. A high connectivity would ensure the space for tissue growth, boost microcirculation of tissue fluid, supply nutrition for the new bones inside the porous scaffold and promote the integration and growth of fibrous tissue and new bones[34].Ultimately, the porous scaffolds should allow the growth of bone tissues and blood vessels.
As shown in Fig. 4(b), the inside walls of the pores are also highly porous. The large pores had sizes at micrometer scale, while the small ones were at nanometer scale, as observed in Fig. 4(c) and (d),respectively. Such porous structures would enlarge the specific surface area of the scaffolds, thus enhancing the absorption of osteogenic proteins,ion-exchange, dissolution-precipitation rate and hence formation of bone-like apatite. Consequently,fibrous tissues are formed, while nutrient transport and waste discharge are promoted[35]. In addition,the wall surface of the pore had a high roughness,leading to strong cell adhesion and enhanced growth of bone tissues[36]. The sample 12# had a porosity of 81.35%, with high connectivity. According to Fig. 4(a), pore size distributions were estimated, as listed in Tab. 4.
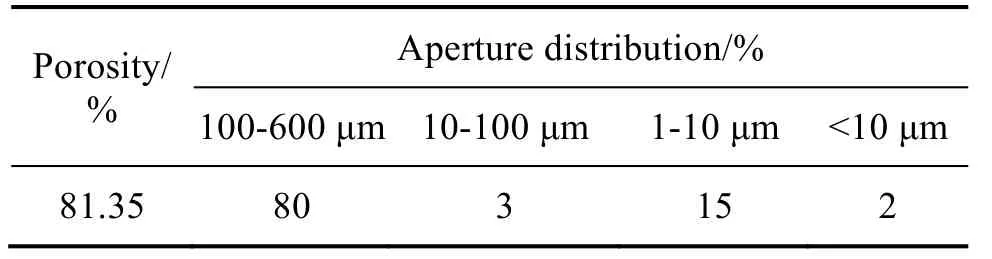
Tab.4 Estimated pore size distributions of the sample 12#
3.5 Mineralization
Fig. 5 shows XRD patterns of the sample 12# before and after mineralization for 10 days.Before mineralization, the sample consisted mainly of HA, CaSiO3, SiO2and Ca2.86Mg0.14(PO4)2. After mineralization in SBF for 10 days, HCA was formed.CaSiO3has promising biocompatibility and is degradable in human body, while it is relatively weak in inducing the deposition of bone-like light apatite[37,38]. HA is a main component in human bones, which could promote the formation of collagen in bones and hence enhance the mineralization, thus having strong biocompatibility and bioactivity[30,40]. SiO2is highly biocompatible[41],while Ca2.86Mg0.14(PO4)2is able to promote the adhesion and increase the reproduction of bone cells, thus demonstrating high cell compatibility and bioactivity[42-44]. The HCA is high compatible with cells[45], further ensuring the growth of bone cells[25].Furthermore, HCA is highly biodegradable[26].
Before mineralization, the average crystal size was 41.5 nm according to Eq. (1), while the maximum size was 65.3 nm. The nanocrystal phases precipitated during the sintering process could helped to increase the mechanical strength of the porous samples. The nanocrystals with different phases had different degradation rates, which was beneficial to the osteoinduction behavior of the scaffolds.

Fig.4 SEM images of the sample 12#: (a, b) surface images, (c, d)zoom-in images of (b)

Fig.5 XRD patterns of the samples 12# before and after mineralization
Fig. 6 shows SEM images of the sample 12#after mineralization in the SBF solution for different time durations. As demonstrated in Fig. 6 (a), after mineralization for 2 days, surface of the sample became smooth, without the presence of any new phase. However, after mineralization for 5 days, the surface morphology was significantly changed.According to XRD patterns, HCA was generated on surface inside the pores of the scaffold. As revealed in the zoom-in image (Fig. 6 (c)) of selected area in Fig. 6(b), the HCA had a fluffy spherical morphology. After mineralization for 10 days, the spherical particles were transferred to leaf-shaped ones, covering the whole surface of the sample, as reflected in Fig. 6 (d). It is then suggested that the sample 12# was of pretty high bioactivity in SBF,thus make it suitable as plant for bone regeneration with strong binding[46,47].
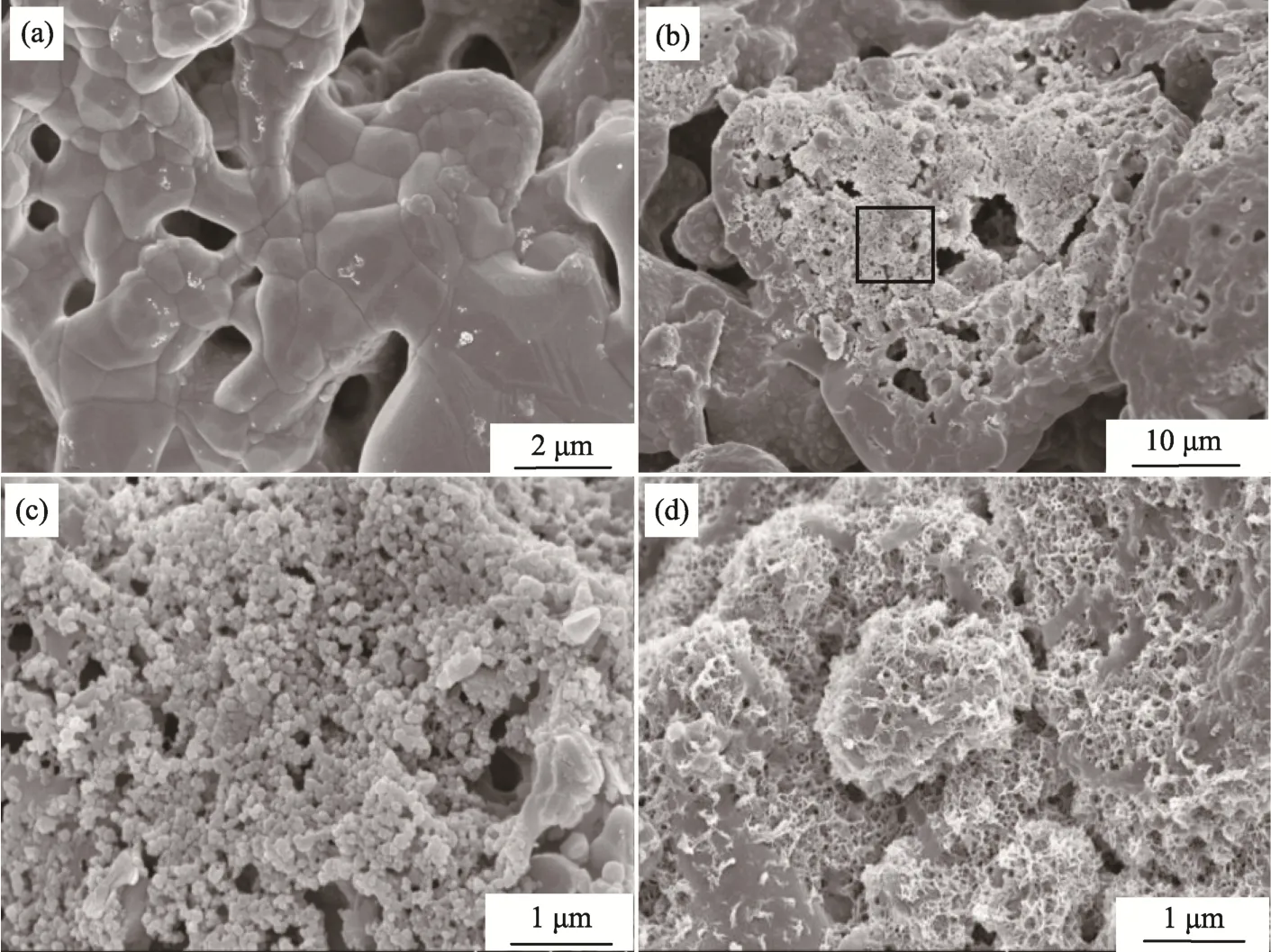
Fig.6 SEM images of the sample 12# after mineralization for different time durations:(a) 2 d, (b) 5 d, (c) partial enlarged picture of panel (b) and (d) 10 d
4 Conclusions
Bioactive porous ceramics, with porosity of 81.35% and flexural strength of 2.75 MPa, could be fabricated by using the foam conversion method from slurries, with the addition of 0.76 wt.% PVA and 30 wt.% lab-made flux, after sintering at 1000 °C. After mineralization for 10 days in SBF solution, a large number of plate-like HAC particles were formed, covering whole surface of the porous ceramic scaffold, showing promising bioactivity. The content of PVA had a significant effect on porosity of the bioactive ceramics, because an appropriate content of PVA resulted in slurries with suitable viscosity to ensure a strong adhesion onto the inside walls of the pores, while not compromising the connectivity of the samples. Due to the presence of millimeter sized interconnected pores in the polyurethane foams and the nanosized pores on the inner walls of the large pores, bone tissues and fibrous vessels could be well grown and the body fluid exchange could be promoted, thus enhancing bone growth.
