
Multi-organ IgG4-related disease continues to mislead clinicians:A case report and literature review
2020-09-10 02:57SandraStrainieneLukasSarlauskasIlonaSavlanValentinaLiakinaIevaStundieneJonasValantinas
World Journal of Clinical Cases 2020年15期
Sandra Strainiene, Lukas Sarlauskas, Ilona Savlan, Valentina Liakina, Ieva Stundiene, Jonas Valantinas
Sandra Strainiene, Ilona Savlan, Valentina Liakina, Ieva Stundiene, Jonas Valantinas, Clinic of Gastroenterology, Nephrourology and Surgery, Centre of Hepatology, Gastroenterology and Dietetics, Institute of Clinical Medicine, Vilnius University, Vilnius 01513, Lithuania
Lukas Sarlauskas, Clinic of Internal Diseases, Family Medicine and Oncology, Centre of Internal Diseases, Institute of Clinical Medicine, Vilnius University, Vilnius 01513, Lithuania
Valentina Liakina, Department of Chemistry and Bioengineering, Faculty of Fundamental Science, Vilnius Gediminas Technical University, Vilnius 10223, Lithuania
Abstract
Key words:IgG4-related disease;Autoimmune pancreatitis;Sialadenitis;Orbitopathy;Autoimmune thyroiditis;Case report;Literature review
INTRODUCTION
Immunoglobulin G4-related disease (IgG4-RD) is a rare systemic chronic fibroinflammatory condition, characterized by tumour-like tissue infiltrations with lymphocytes and IgG4-secreting plasma cells, storiform fibrosis and often, but not always, elevated serum levels of IgG4, resulting in organ swelling, scaring and dysfunction[1-3].The disease can affect almost every organ, usually pancreas, biliary ducts, salivary glands, orbits, kidneys, and lymph nodes[1,2,4].The systemic nature of this disease has been recognized only in the 21stcentury by Hamanoet al[5,6]in Japan.Despite the growing recognition of this new condition, adequate epidemiological data,and unified evidence-based guidelines for IgG4-RD diagnosis and management are still lacking.Our knowledge about IgG4-RD prevalence and manifestation in Europe and other Western countries is limited, as the majority of clinical data analysis has been performed in Asia, mostly Japan[7,8].The diagnostic process of IgG4-RD is challenging, as it takes time and referrals to many different specialists to establish the correct diagnosis.
We report a case of IgG4-RD manifested as thyroiditis, orbital myositis, orbitopathy,uveitis, scleritis, sialadenitis, autoimmune pancreatitis (AIP) and prostatitis in a 55-year-old man.It is the first presented case from Lithuania as well as from the Baltic Sea region.Our goal is to take a closer look at this rare disease and its manifestations,differential diagnosis and management, according to the latest evidence-based data.
CASE PRESENTATION
Chief complaints
A 54-year-old Caucasian male was admitted to the Centre of Hepatology,Gastroenterology and Dietetics presenting with complaints in various organ systems.The patient was complaining of fatigue, weight loss (lost over 30 kg in 9 mo despite good appetite), frequent bowel movements (up to 3 times daily), dryness of the mouth along with changes of facial appearance due to bilateral salivary gland swelling, and difficulty urinating.
History of past illness
At the age of 36, the patient was diagnosed with autoimmune thyroiditis.It was managed with a low dose of prednisolone.The disease re-occurred as subacute thyroiditis several times within 15 years.This resulted in hypothyroidism, and 50 mcg of levothyroxine was added.The patient also had bronchial asthma, which was well controlled with bronchodilators on demand and currently in remission.Whereas the patient had a familial anamnesis of the thyroid disease, no additional testing for other possible reasons of thyroid disease was made at that time.
At the age of 52, the patient approached the ophthalmologist with a swollen right eye and visual impairment.The treatment with anti-inflammatory ointments,antibiotics and corticosteroid eye drops was initiated.However, the symptoms progressed, and the patient developed diplopia and strabismus, which did not respond to the vision correction with prism lenses.The neurologist then consulted the patient to differentiate possible mass of thesella turcica.Magnetic resonance imaging(MRI) of the head showed no pathology.Though the patient's symptoms further progressed and he developed the significant protrusion of both eyes, photophobia,eye-watering and impaired diplopia.At this point, the neurologist and ophthalmologist repeatedly consulted the patient.Negative antibodies against acetylcholine and neuromyography excludedmyasthenia gravis.Ultrasound examination of the extracranial cerebrovascular system showed nosinus cavernosuspathology or carotid artery stenosis.Additionally, the inconsistent size heterogenic mass was spotted transorbitally on the left orbital cavity (Figure 1).
The 1.5-T MRI scan of the orbital cavity pointed out the substantial enlargement in extraocular muscles (left lateral and inferior rectus muscles, right superior rectus muscle), sinusitis, and no signs of lacrimal glands pathology (Figure 2).The symptoms were attributed to endocrine ophthalmopathy (Graves' disease), and the patient was referred to the tertiary Centre of endocrinology.At the time of admission, the patient’s laboratory tests showed normal levels of thyrotropin hormone (TTH) at 1.28 mU/L(normal range 0.4-4.0 mU/L), free thyroxine (FT4) 18.71 pmol/L (normal range 12–22 pmol/L) and anti-thyroid peroxidase (anti-TPO) antibodies at 0.6 kU/L (normal range<5.61), also negative thyrotropin receptor antibodies (TRAb) at 5.1 U/L (negative when <9).The patient then received twelve courses of weekly intravenous methylprednisolone (in total 4.5 g of methylprednisolone).Although the periorbital swelling decreased, diplopia, photophobia, and tearing remained.The patient then received a course of orbital radiotherapy for steroid-refractory endocrine ophthalmopathy.However, this treatment was also not sufficient enough.The patient was referred to otolaryngologist regarding the indications for the left orbital decompression due to aggressive exophthalmos.The surgical treatment was not indicated, though.The endocrinologists then doubted the diagnosis of Graves' disease and thought of idiopathic orbital inflammation, which can be treated with retrobulbar steroid injections.However, the patient refused this treatment after several injections and was out of reach for a year.
Two years later, the patient started complaining of abdominal pain, diarrhoea,severe weight loss (lost 20 kg per half-year), fatigue and urinary problems.Internal medicine doctor referred him to the gastroenterologist.Ultrasound of the abdomen showed hepatosteatosis, gastroscopy–erosive gastropathy, duodenal biopsy specimens revealed no signs of the celiac disease and colonoscopy indicated tubular adenoma.The patient had a urologist's consultation due to urinary hesitancy which was attributed to benign prostatic hyperplasia.At this period, the patient was also diagnosed with diabetes, which was managed with a strict diet and metformin.
As the patient developed vitiligo, muscular pain, sicca symptoms and severe fatigue, he was sent to the rheumatologist, and multiple blood tests were performed.Positive IgG, A, M antibodies against β-2 glycoprotein-1 and cardiolipin, positive antinuclear antibodies (ANA), negative anti-neutrophil antibodies (ANCA) and negative extractable nuclear antigens (ENA) (including anti-SSA, anti-SSB) were detected.Rheumatoid factor, complement C4 and C3c were within a normal range(Table 1).These findings did not confirm any systemic rheumatological disease.However, elevated liver enzymes were noticed, and the patient was referred to the Centre of Hepatology, Gastroenterology and Dietetics for the liver biopsy.
Family and personal history
The patient had had bronchial asthma since childhood, which was currently in remission.His mother underwent surgery on her thyroid gland in her youth, and his sister had second type diabetes.The patient was a non-smoker and denied alcohol consumption.He claimed to have no allergies to food or medicines.

Table 1 Immunological tests
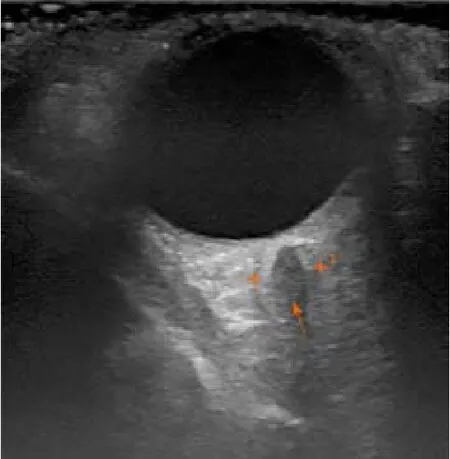
Figure 1 Orbital sonography.
History of present illness
Patient's symptoms started in his youth and had been continuously worsening and spreading to different organs throughout the years.The most recent deterioration started about 9 mo ago when he began losing weight, complaining of diarrhoea and severe fatigue, sicca symptoms.Several weeks before the hospitalization, he noticed continuously increasing masses on the neck, severe dryness of the mouth, difficulty swallowing and urinating.
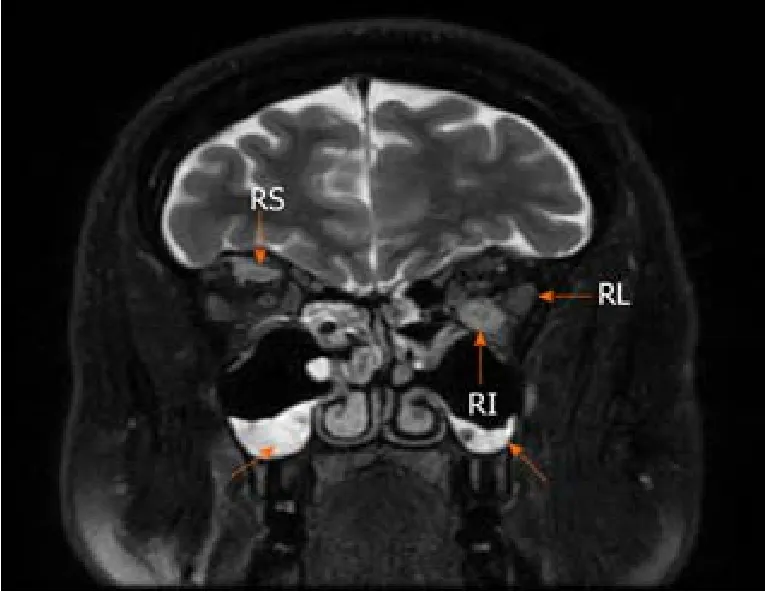
Figure 2 Magnetic resonance imaging of the orbital cavity.
Physical examination
At presentation, the patient was asthenic (height 188 cm, weight 68.3 kg, body mass index 19.32 kg/m2) and had vitiligo-like patches on the insides of his wrists and the lower left calf.We also noted large palpable hard masses in the projection of submandibular and parotid salivary glands bilaterally, diffuse I B stage goitre and the protrusion of the left eyeball.
Laboratory examinations
Blood analysis was almost within the normal range (Table 2).There were no elevated liver enzymes or signs of liver dysfunction, viral hepatitis markers were negative.
Imaging examinations
As there were no indications for liver biopsy, it was decided to perform abdominal MRI which displayed changes of pancreatic parenchyma, showing diffusely enlarged sausage-shaped pancreas with a peripancreatic rim of edematous tissue that is pathognomonic for AIP (Figure 3).Severe weight loss with diarrhoea and diabetes indicated the endocrine and exocrine pancreatic insufficiency.
Further diagnostic work-up
Combination of AIP, orbital myositis, salivary gland involvement, as well as the previous diagnosis of thyroiditis and uveitis, increased the suspicion of the IgG4-RD.Differential diagnosis was kept as possible Sjögren's syndrome (SS), lymphoma, and sarcoidosis.Consequently, we performed the ultrasound imaging of the neck.The thyroid was normal in size with several nodules on the right and left lobes (8 mm × 13 mm and 7 mm × 21 mm) (Figure 4A and B).There were several symmetrical rounded parenchymal masses (28 and 33 mm) in the area of submandibular glands(Figure 4C and D) and altered parotid gland tissue with indeterminate size masses observed bilaterally.It was decided to perform fine-needle aspiration from the palpable masses in the neck.
Histopathological examination of the salivary gland biopsy revealed signs of chronic lymphoepithelial sialadenitis:The infiltration of T and B lymphocytes,moderate plasma cell and eosinophil infiltration, acinar atrophy, moderate fibrosis.However, no clear histological and immunohistochemical evidence of IgG4-mediated chronic sclerotic sialoadenitis was found.There was no deposit of amyloid, no granuloma, any signs of mucosa-associated lymphoid tissue lymphoma or Langerhans histiocytosis.We also found elevated plasma IgG4 concentration at 6.87 g/L (normal range 0.08-1.40 g/L).
MULTIDISCIPLINARY EXPERT CONSULTATION
Given the multiple organ involvement, high IgG4 levels, the initial findings on the MRI scan and salivary gland biopsy, the diagnosis of the IgG4-RD presented with thyroiditis, orbital myositis, orbitopathy, uveitis and scleritis, sialadenitis, AIP and prostatitis was confirmed at the multidisciplinary expert consultation by the gastroenterologist, endocrinologist, rheumatologist, opthalmologist and radiologist.It was discussed whether the patient's asthma is a pulmonary involvement of IgG4-RD.As it started in childhood and was in remission, it was decided that bronchial asthma is a co-morbid atopic disorder.The origin of the patient's urination problems remained questionable, but the majority of consultants voted for chronic IgG4-related prostatitis.The need to perform an additional biopsy from another involved organ was also intensely debated.A decision was made, that there were enough criteria to diagnose the IgG4-RD, and the biopsy result would not affect the course of treatment.
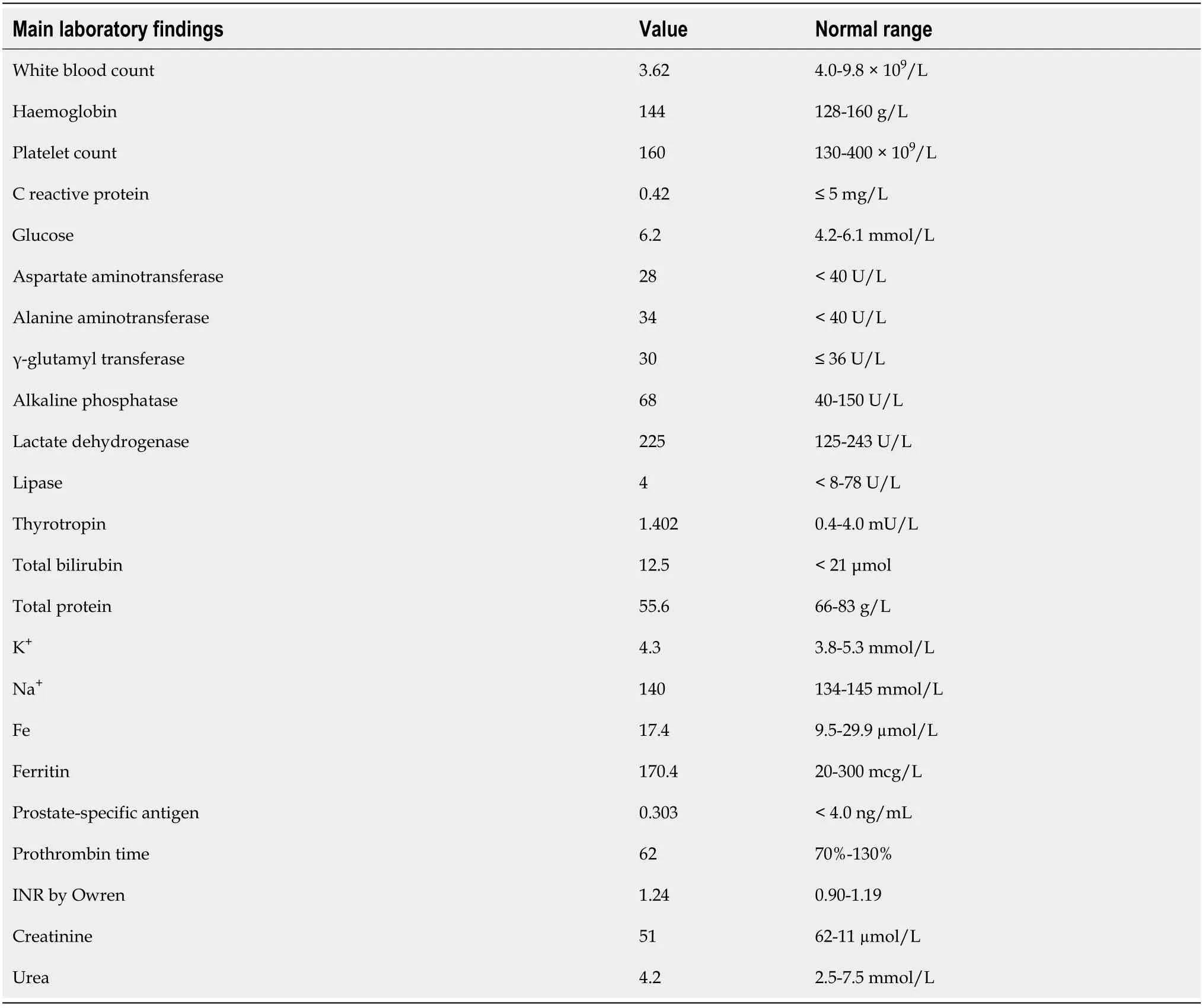
Table 2 Main laboratory findings
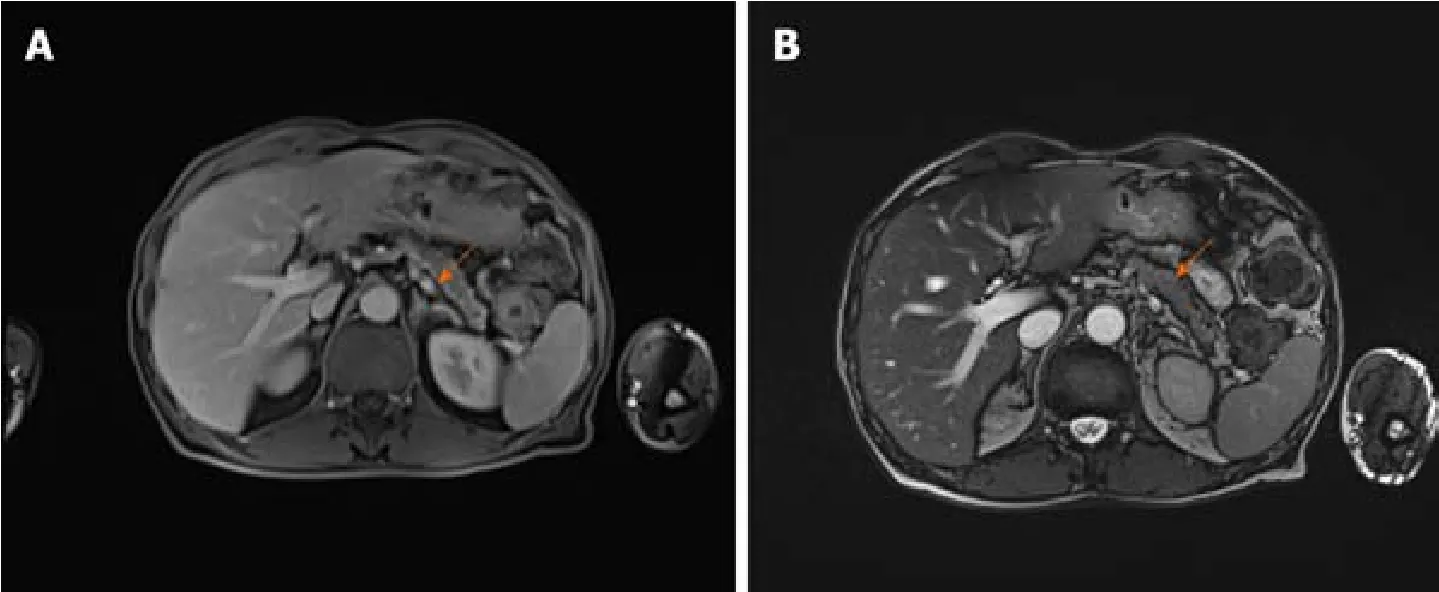
Figure 3 Abdominal magnetic resonance imaging before the treatment.
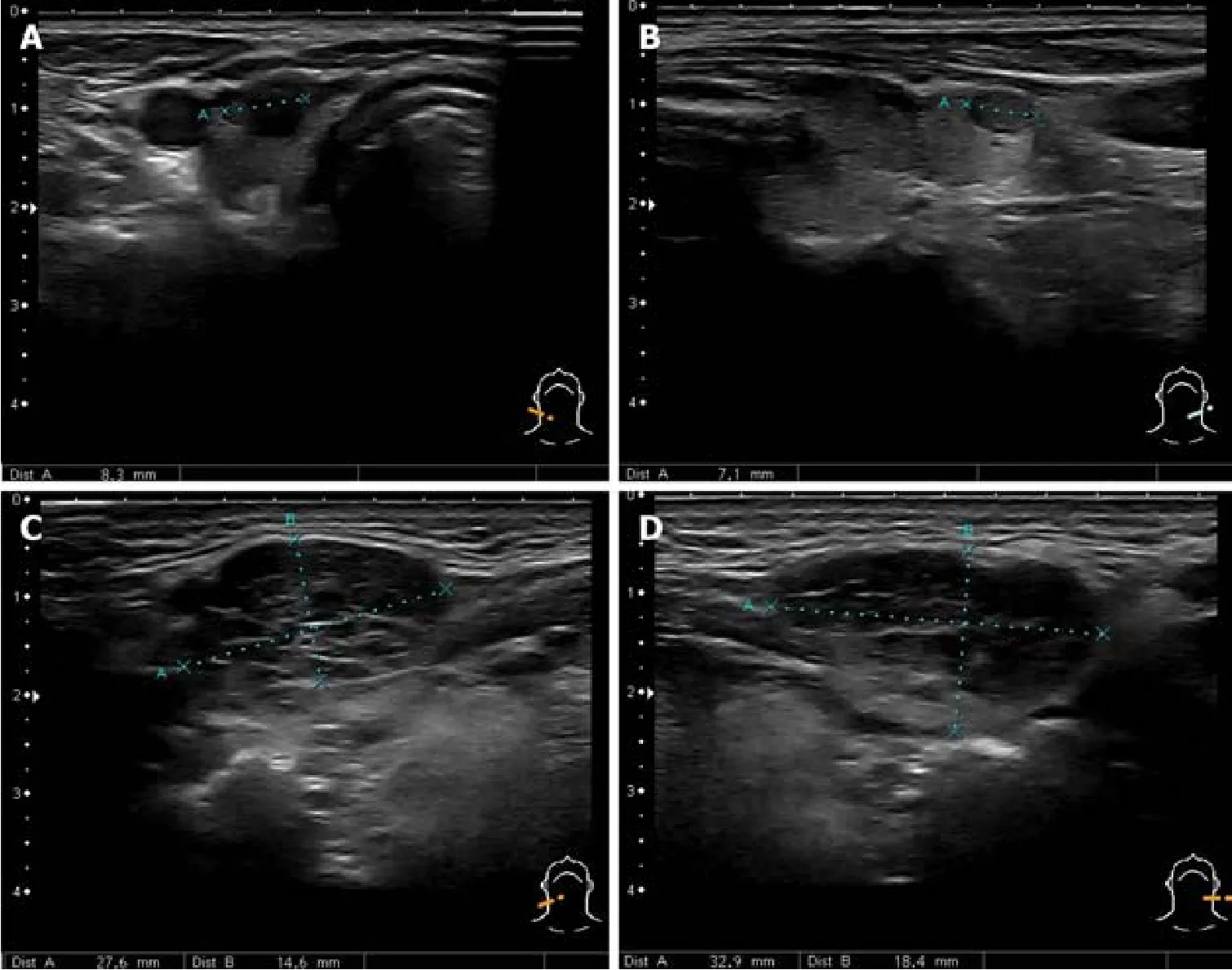
Figure 4 Ultrasound of the neck.
The treatment with an initial dose of 0.6 mg/kg/d of prednisolone was recommended, and the follow-up visit and MRI were scheduled after one month.In case of non-response, it was decided to considerate treatment with rituximab.
FINAL DIAGNOSIS
The final diagnosis of the case was IgG4-RD presented with thyroiditis, orbital myositis, orbitopathy, uveitis and scleritis, sialadenitis, AIP and prostatitis.
TREATMENT
The induction therapy with 40 mg/d (0.6 mg/kg/d) of prednisolone for 4 wk was started immediately and tapered until the maintenance dose of 5.0 mg/d.
OUTCOME AND FOLLOW-UP
A follow-up visit one month later revealed significantly shrunken masses in front of the ears and the several times decreased serum IgG4 concentration (from 6.87 g/L to 1.93 g/L).Repeated MRI showed a significant response to glucocorticosteroid (GC)therapy with no signs of pancreatitis.(Figure 5).A year after the diagnosis and treatment, the patient's overall condition significantly improved:The majority of symptoms regressed, and he gained back most of his lost weight.Diabetes was under control without any antidiabetic drug use, urination problems resolved.Exophthalmos also improved over time.The grade of left proptosis decreased, however, diplopia,vision disturbance and strabismus remained.The recession of the left inferior rectus muscle was recently performed to correct the high-level strabismus.
The patient is now on maintenance therapy of 5 mg of prednisolone.Control MRI 16 months later showed no signs of pancreatitis or other organ abnormalities.The patient is now carefully followed up every three months by the gastroenterologist,endocrinologist and ophthalmologist.
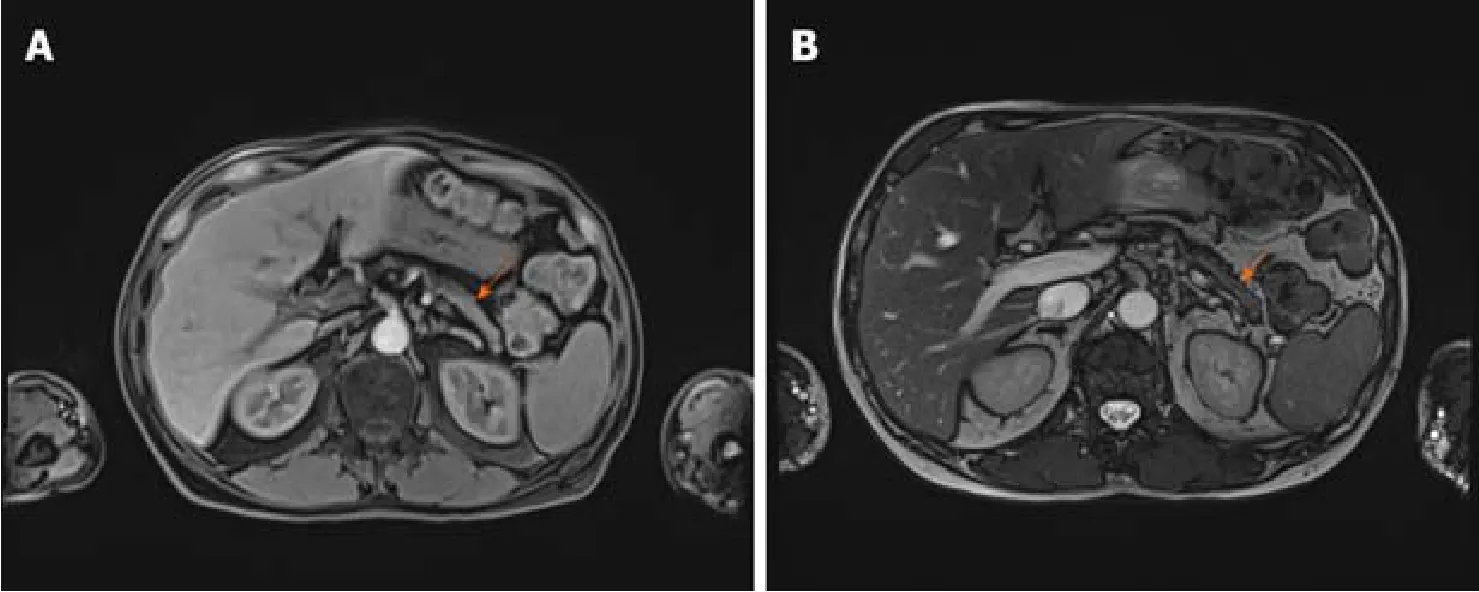
Figure 5 Abdominal magnetic resonance imaging after the treatment.
DISCUSSION
The exact prevalence of IgG4-RD is unknown, as the condition still does not have its own unique International Statistical Classification of Diseases -10 code and the International Classification Criteria have been validated just recently[9].According to the research carried out in Japan, the incidence of the IgG4-RD is 0.28-1.08/100000 and 336-1300 patients are newly diagnosed per year[7].The extent of IgG4-RD in Europe and Northern America is still unknown and might be underestimated[3,9].There are only several multicentric studies of IgG4-RD in Europe conducted[10-12].
The existing studies suggest that IgG4-RD predominantly affects middle-aged and older men (50-60 years).It was noticed that AIP, sclerosing cholangitis, retroperitoneal fibrosis, and tubulointerstitial nephritis are more common in men, while women are more often diagnosed with sialadenitis, dacryoadenitis and thyroiditis[13].In our case,the patient was a male diagnosed with AIP as well as sialadenitis and thyroiditis.
Depending on the pattern of affected organs, clinicians attempt to classify IgG4-RD.The best-known four phenotypes of IgG4-RD were created by Wallaceet al[14]The authors distinguished four possible IgG4-RD subgroups:Pancreato-hepatobiliary disease, retroperitoneal fibrosis and/or aortitis, head and neck-limited disease and classic Mikulicz syndrome with systemic involvement.The evolving recognition of the disease determines new possible classifications.Recently,Niwamotoet al[15]suggested the fifth group, which includes AIP and other systemic involvement.This subgroup turns out to be the most suitable in our case, as it includes dominant lesions of the pancreas, salivary glands and orbits.
IgG4-RD diagnosis
The presentation of IgG4-RD is typically subacute or chronic, marked by a spectrum of mild localized symptoms to significant tissue damage and organ failure evident for months or even years before diagnosis complicating the recognition of the disease[1,2,4,8].
The main diagnostic patterns for IgG4-RD are clinical symptoms, histological findings, elevated IgG4 levels and typical radiological findings.Until now, there have been two types of diagnostic criteria available:The Mayo Clinic HiSORt criteria for AIP and the Japanese Comprehensive Clinical Diagnostic criteria for IgG4-RD[16,17].The American College of Rheumatology (ACR)/European League Against Rheumatism(EULAR) Classification Criteria for IgG4-RD were finally developed and validated at the end of 2019[9].A case meets the classification criteria for IgG4-RD, if the entry criteria are met, no exclusion criteria are present, and the total points are ≥ 20[9].
The main histopathological features of IgG4-RD are:(1) Dense lymphoplasmacytic infiltration with increased numbers of IgG4 positive plasma cells and often increased eosinophils;(2) Storiform-type fibrosis;and (3) Obliterative phlebitis[1].However, these findings may not be present within lacrimal and salivary glands, lymph nodes, and interstitial lung disease[1,18].
Elevated serum IgG4 (>1.35 g/L) concentration is useful in suspecting IgG4-RD,monitoring the disease activity and predicting relapse[2,17].Another possible biomarker of the disease might be the increased circulating plasmablast counts in the blood[19].Imaging is usually the first diagnostic tool to identify and differentiate the tumorous swellings throughout the body that occur in patients with IgG4-RD.Some of them are pathognomonic, such as in our case when MRI showed pancreatic enlargement with"sausage-shaped" appearance and a low-attenuating capsule-like "rim" around the enlarged pancreas.
In our case, the diagnosis was established based on the comprehensive criteria(clinical presentation, typical radiological findings, elevated IgG4 concentration).Our patient also met all the newly established ACR/EULAR criteria (31 points).The histopathological examination from the salivary gland was not specific and showed chronic lymphoepithelial sialadenitis, which could be the manifestation of SS or viral infection.
Diagnostic challenges of the presented case
IgG4–RD can mimic various malignant, infectious or other inflammatory conditions.In our case, symptoms progressed around 18 years metachronously affecting different organs.The patient was treated at several departments, had multiple appointments to eight specialists (endocrinologist, ophthalmologist, neurologist, otolaryngologist,rheumatologist, urologist, internal medicine doctor and gastroenterologist) due to different symptoms, which later turned out to be the manifestations of IgG4-RD.The delay to diagnosis between the inial symptoms and the discovery of elevated IgG4 blood levels or typical histologic findings variates between 5 ± 11.2 years[20].The main problem in our case was the lack of a comprehensive approach to the patient as his symptoms presented and were managed as unrelated disorders.The majority of specialists looked at only one organ system disregarding the whole picture.
Firstly, the disease affected the thyroid gland, and autoimmune thyroiditis was diagnosed.Although autoimmune thyroiditis usually affects women, the patient was not investigated for other possible causes of thyroid dysfunction, as he had familial anamnesis of thyroid disease.However,hypothyroidism can be the first sign of IgG4-related thyroiditis (also called Riedel's thyroiditis and a fibrosing variant of Hashimoto's thyroiditis)[2,4].The first episode of eyes involvement was addressed to simple conjunctivitis.While the patient gradually developed protrusion of both eyes,photophobia and severe diplopia, the ophthalmologist and the neurologist wandered betweensella turcicamass and myasthenia gravis.However, orbital MRI showing signs of orbitopathy, brought the patient back to the endocrinologist who diagnosed endocrine ophthalmopathy based on the previous anamnesis of autoimmune thyroiditis and provided the treatment plan with 12 wk hospitalizations for intravenous GC pulse therapy following the radiotherapy.As the treatment was not effective, the endocrinologists then doubted the diagnosis of Graves' disease and considered of idiopathic orbital inflammation, which, according to the literature, can be treated with retrobulbar steroid injections[21].However, it was not then thought of the relation with IgG4-RD at that time.As the patient was out of reach for a year after refusing to continue this treatment, he was not further examined for the possible systemic disease at that time.Retrospectively the patient's symptoms can be attributed to the development of IgG4-related orbital myositis, uveitis and scleritis.Proptosis and exophthalmos were a result of orbital pseudotumours that involved extraocular muscles (orbital myositis).Photophobia, constant tearing and visual impairment can be attributed to scleritis and uveitis, which are the less common ophthalmic manifestations of IgG4-RD[18,22].
Afterwards, the patient experienced severe weight loss, diarrhoea and abdominal pain.These symptoms led the patient to the gastroenterologist.Examination for possible celiac disease, inflammatory bowel diseases and cancer was performed,though none of these diseases was confirmed.The suspicion of the systemic disease increased when the patient developed vitiligo like patches, sicca symptoms and muscle pain.The rheumatologist consulted and investigated the patient, yet none systemic connective tissue illness was confirmed.
Incidentally, the elevated liver enzymes were observed, and the patient returned to the gastroenterology department for liver biopsy.Instead, the needle-aspiration from significantly enlarged palpable salivary gland was performed.The considerations in the differential diagnosis included Sjögren's syndrome, granulomatosis with polyangiitis, sarcoidosis, lymphoma, infection, Graves' thyrotoxicosis and cancer.AIP on abdominal MRI linked diarrhoea, weight loss and impaired glucose tolerance to the signs of exocrine and endocrine pancreatic failure.Sialadenitis with changes in facial appearance is particularly characteristic of IgG4-RD[2,4,23].In our case, the enlargement of submandibular and parotid salivary glands accompanied by AIP, orbital myositis,as well as the previous diagnosis of thyroiditis, were the key symptoms for suspecting the IgG4-RD and leading clinicians out of the woods (Figure 6).As we confirmed the diagnosis of IgG4-RD, it was then considered thoroughly on the final determination of affected organs.The main questionable issues were pulmonary and urogenital involvement.It was discussed whether the patient's asthma is not a pulmonary involvement of IgG4-RD.According to the literature, asthma might be both, a part of pulmonary involvement and atopic process as well[8].A history of atopic disease,including bronchial asthma, allergic rhinitis, nasal polyps, and atopic dermatitis were reported in approximately one-third of IgG4-RD patients[24].The pulmonary involvement of IgG4-RD usually manifests as adult-onset, refractory and difficult-totreat asthma-like symptoms (cough or wheezing)[25,26].Our patient was diagnosed with bronchial asthma in childhood, and it was well-controlled with only a rare on-demand need of bronchodilators and currently in remission.Therefore, it was decided that bronchial asthma is a separate co-morbid disorder.
The origin of the patient's urination problems was also questionable at first.Patients with IgG4-RD often complain of frequent urination and feelings of residual urine,which are same as the symptoms of benign prostatic hyperplasia[27].These symptoms totally regressed after initial treatment with prednisolone.Therefore, there was no doubt of IgG4–related prostatitis.
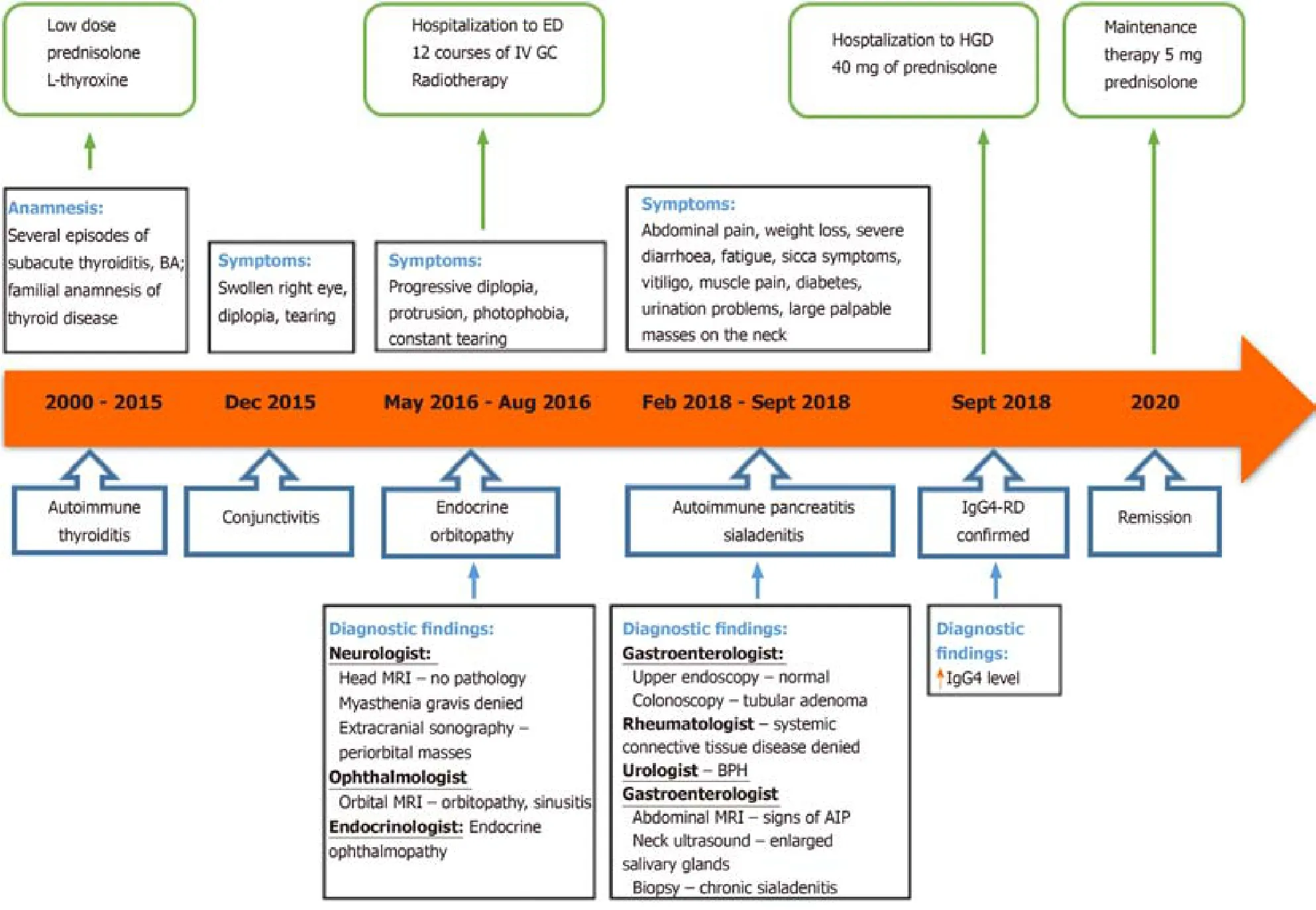
Figure 6 Case history timeline.
IgG4-RD treatment
The treatment option for patients with IgG4-RD is based on clinical experience and expert opinion, as there are no controlled or prospective studies for the best treatment approach[3,18].The biggest challenge of IgG4–RD is to determine the best treatment strategy.To date, most clinical manifestations of IgG4-RD respond well to initial induction therapy with 0.6 mg/kg/d or fixed 30-40 mg/d dose of prednisolone for four weeks and then tapering by 2.5-5 mg/wk[4,28-30].In case of good response, there are two possibilities:Taper GC until patients are off the drugs or until the maintenance dose of 2.5-5.0 mg/d which is continued from 6 months up to 3 years[2,4,31].The use of the higher doses of GC are reported in patients with severe complications (pancreatic,pulmonary, renal and retroperitoneal involvements), and lower doses are suggested in patients with cardiovascular diseases and the elderly[31].Long-term maintenance therapy with low-dose GC (2.5-5 mg/d prednisolone) can be applied for patients with a higher risk of relapse[18,28].Rates of response to the GC therapy are 79%-97%, whereas 31%-60% of patients tend to relapse[4].
Immunomodulators (IM) (mostly 2.0-2.5 mg/kg/d of azathioprine) are used for the maintenance of the remission in relapsing/complicated IgG4-RD or as steroid-sparing agents[2,4].
Another effective drug for the management of IgG4-RD is a B–cell depleting agent–rituximab (RTX), which is a viable option for induction and maintenance of the disease in cases of GC intolerance, incomplete resolution, requiring ≥ 20 mg/d of prednisolone, or IMs intolerance;also the advanced disease spread to multiple organs[2,4,32,33].
Currently available therapies for IgG4-RD, including GC and RTX, are associated with many potential side effects, and their long-term use can get complicated as the disease mostly affects middle-aged to elderly individuals.Advances in understanding the disease pathogenesis have given some promising ideas which may lead to new therapeutic options, such as bortezomib, abatacept and dupilumab[34-38].Infliximab was recently proposed for the treatment of IgG4-related orbital disease refractory to GC[35,39].
In our case, the initial treatment with prednisolone was effective.Within a month,there was a favourable clinical response:The tumefactive masses shrunk significantly,serum IgG4 level decreased several times, and there were no signs of pancreatitis on MRI.As there is a high risk of relapse due to multiple organ involvement, the patient is still on the long-term maintenance therapy with 5 mg/d of prednisolone.
IgG4-RD Responder Index
IgG4-RD responder index (RI) created in 2012 by Carruther'set al[40]is the only currently available objective tool for the evaluation of a patient's disease activity that could be used to assess treatment response, though still in development.We also applied the IgG4-RD RI tool to our case.The patient demonstrated a striking clinical improvement within one month after starting prednisolone.IgG-RD RI decreased from 11 to 4.
CONCLUSION
Due to the heterogenicity of the disease, the IgG4-RD diagnosis is a challenge to every clinician, therefore, usually is delayed.We report the first clinical case of multi-organ IgG4-RD from Lithuania and the Baltic Sea region, which highlights the importance of a comprehensive approach to the patient in order to achieve effective recognition of the rare immune system malfunction conditions such as IgG4-RD.To date, the treatment with GC significantly improves patients' condition and quality of life.However, more extensive studies are needed to maintain a worldwide accepted evidence-based diagnostic and treatment guidelines for clinical use.Further epidemiological studies, especially in the Western World, are lacking.
ACKNOWLEDGEMENTS
We wish to thank the patient and Vilnius University Hospital Santaros Clinics for giving the consent and providing the data to report this case.
 World Journal of Clinical Cases2020年15期
World Journal of Clinical Cases2020年15期
- World Journal of Clinical Cases的其它文章
- Impacts and challenges of United States medical students during the COVID-19 pandemic
- Recent advances in the management of gastrointestinal stromal tumor
- Medical research during the COVID-19 pandemic
- Progress of intravoxel incoherent motion diffusion-weighted imaging in liver diseases
- Typical and atypical COVID-19 computed tomography findings
- Review of possible psychological impacts of COVID-19 on frontline medical staff and reduction strategies