
巴洛沙韦酯(Baloxavir Marboxil)合成路线图解
2020-09-01 06:44李召辉
山东化工 2020年15期
张 铮,李召辉,袁 杰
(1.北大医药股份有限公司,重庆 400714;2.中国科学院大学 重庆仁济医院 药剂科,重庆 400062)
巴洛沙韦酯(Baloxavir Marboxil),化学名({( 12aR)-12-(11S)-7,8-二氟-6,11-二氢二苯并[b,e]硫-11-基]-6,8-二氧代-3,4,6,8,12,12a-六氢-1H-[1,4]噁嗪[3,4-c]并吡啶[2,1-f][1,2,4]三嗪-7-基}氧代)甲基碳酸甲酯,CAS登记号为1985606-14-1。巴洛沙韦酯为前体药物,经肠道上皮、肝脏和血液中的酯酶水解,迅速转化为活性形式Baloxavir。Baloxavir抑制流感病毒帽状依赖型核酸内切酶(cap-dependent endonuclease,CEN),从而抑制病毒RNA的合成。巴洛沙韦酯由日本盐野义(Shionogi)制药株式会社和瑞士罗氏(Roche)公司合作开发,2018年2月在日本上市,同年10月在美国上市,商品名Xofluza,用于单剂量治疗症状不超过48小时的急性、无高危因素的成人和12岁或以上的青少年甲型和乙型流感患者[1-4]。
巴洛沙韦(2)是用苄基保护的中间体B或正己基保护的中间体C与中间体A缩合,经脱保护后与氯甲基乙基碳酸酯(3)反应得到巴洛沙韦酯(1)。中间体A、B、C各有一个手性中心。本文对巴洛沙韦酯(1)及中间体A、B、C的合成路线进行综述,以找到适合工业化生产的合成路线。
1 中间体A的合成路线
1.1 以3,4-二氟苯甲酸(2a)为起始物料
在-40℃条件下2a 用二异丙基胺基锂处理后发生2位金属化,与DMF反应生成醛,再分子内环合生成3a;3a在D-樟脑磺酸的催化下与苯硫酚(4a)反应,得到5a,然后用1,1,3,3-四甲基硅氧烷和三氯化铝还原生成6a;6a在多聚磷酸(PPA)中环合得到25a,再用硼氢化钠还原得到中间体A。该路线共五步,总收率71%[5]。该路线第一步用到二异丙基胺基锂,反应需无水并在-40℃进行,条件苛刻,第二步用到苯硫酚,刺激性气味重,工业化难度较大。
1.2 以1,2-二氟-3-甲基苯(7a)为起始物料
7a在三氯化铝催化下与液溴发生溴代反应得到8a;8a与异丙基氯化镁发生格氏交换,再与二氧化碳反应得到9a;在偶氮二异丁腈催化下与NBS发生溴代反应得到10a;10a与苯硫酚钠(11a)反应得到6a,然后经环合、还原得到中间体A[6]。该路线共六步,总收率49.4%,用苯硫酚钠代替了刺激性气味重的苯硫酚,但需用到毒性和腐蚀性很强的液溴和易燃易爆的偶氮二异丁腈,不适合工业化。
1.3 以 2-溴甲基-3,4-二氟苯乙腈(12a)为起始物料
12a经苯硫酚钠(13a)取代得到14a;14a在五氧化二磷作用下环合得到25a,随后还原得到中间体A[7]。该路线共三步,总收率80.9%,该路线较短,但起始物料12a不易获得。
1.4 以(6-溴-2,3-二氟苯基)甲醇(15a)为起始物料
15a与氢溴酸发生取代反应得到16a;在惰性气体保护下二苯二硫醚(17a)经硼氢化钠处理后与16a反应得到18a;在惰性气体保护和-35℃的条件下,18a与异丙基氯化镁发生格氏交换,再与二氧化碳反应得到19a;19a经傅克酰基化反应合成得到25a,然后还原得到中间体A[8]。该路线共五步,总收率34.6%,该路线用二苯二硫醚替代苯硫酚,但第二步用到对水和空气敏感的异丙基氯化镁,反应条件苛刻。
1.5 以3,4-二氟-2-甲基苯甲酸(20a)为起始物料
20a和硫酸二甲酯发生酯化反应得到21a,再与NBS发生溴代反应得到22a;在三氯化铁和锌粉的存在下,22a与二苯二硫醚(17a)反应得到23a,然后经水解、环合与还原得到中间体A。该路线共六步,总收率75%,纯度98.04%[9]。该工艺避免了路线1.1的苛刻反应条件,并用二苯二硫醚替代苯硫酚,适合工业化生产。
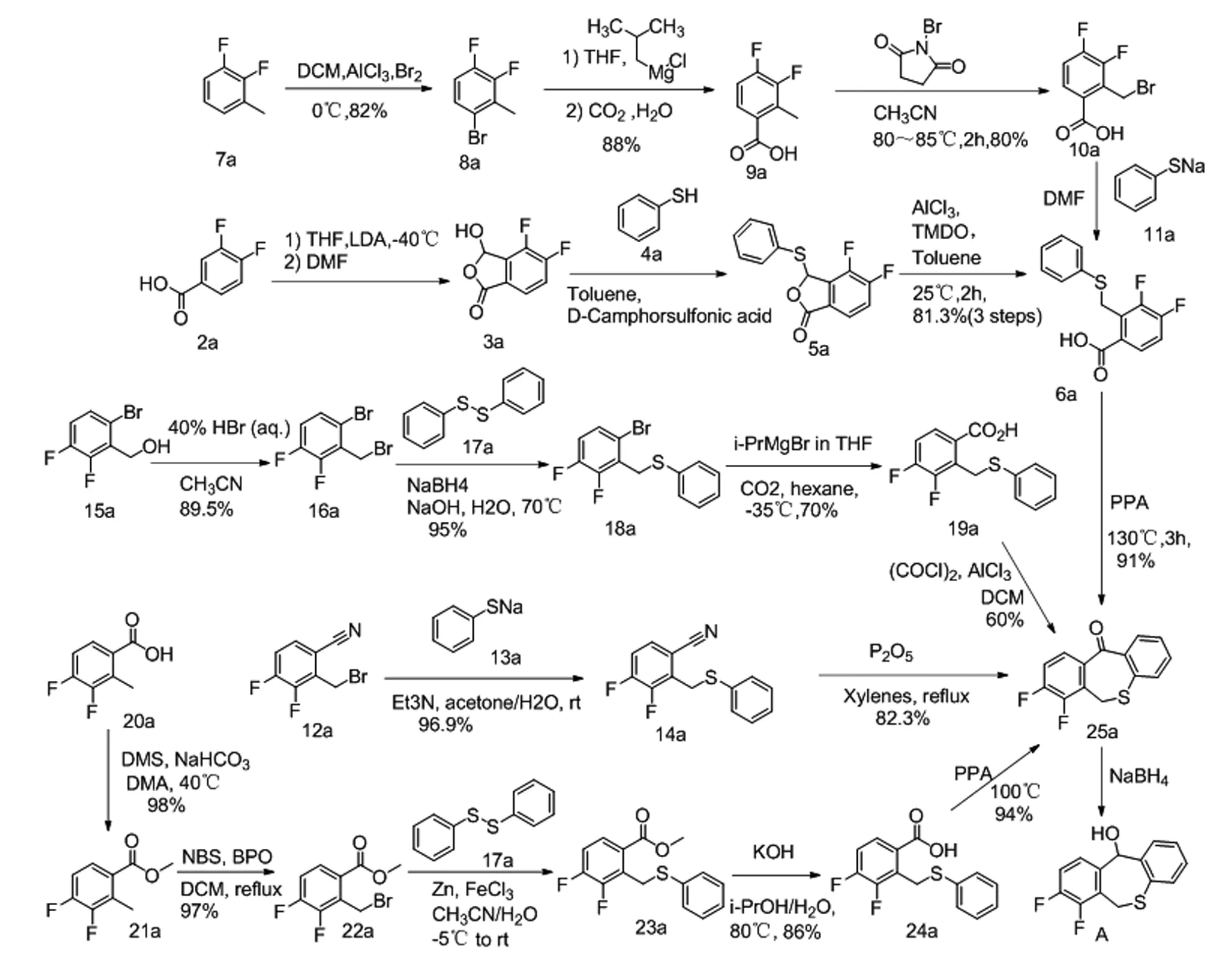
图1 巴洛沙韦酯中间体A合成路线
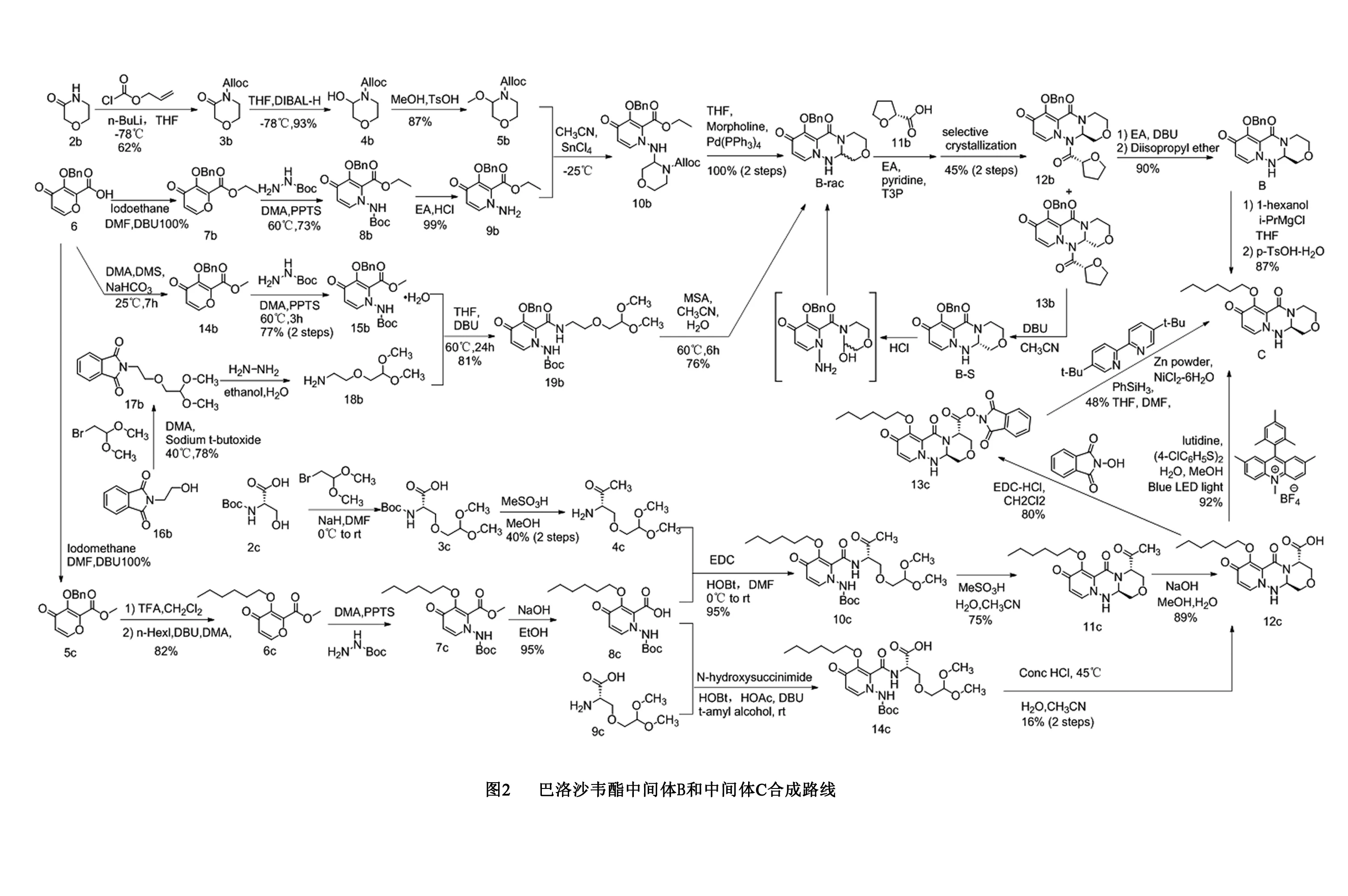
2 中间体B的合成路线
2.1 路线一
在-78℃及正丁基锂存在下,3-吗啉酮(2b)用氯甲酸烯丙酯保护,然后在-78℃用二异丁基氢化铝还原得到4b,在酸性条件下4b与甲醇形成甲氧基化合物5b;3-(苄氧基)-4-氧代-4H-吡喃-2-羧酸(6)在碘乙烷发生酯化反应得到(7b),然后与Boc-肼反应得到8b,再脱去Boc保护基团得到9b;在-25℃及四氯化锡存在下,片段5b与片段9b连接得到10b,随后经脱保护和环合得到外消旋体B-rac[10]。
B-rac与(R)-四氢呋喃-2-羧酸(11b)缩合生成非对映异构体12b和13b,经选择性结晶得到(R,R)-非对映异构体(12b),然后脱去四氢呋喃甲酰基,用二异丙醚结晶得到中间体B[10]。该路线以6为起始物料制备中间体B,共8步,总收率29%。该路线在吗啉酮片段的构建中使用到-78℃的低温条件,且正丁基锂和二异丁基氢化铝对水和空气敏感,工业化难度较大。
2.2 路线二
N-羟乙基邻苯二甲酰亚胺(16b)与2-溴-1,1-二甲氧基乙烷反应制备17b,再脱保护得到18b;6与硫酸二甲酯反应得到14b,然后与Boc-肼反应得15b;片段18b与15b在DBU催化下发生酰胺化反应得到19b,然后用甲磺酸处理19b得到B-rac。按照路线一的方法进行拆分,得到中间体B[5]。该路线以6为起始物料共7步,总收率19%,该路线采用拆分的方法制备中间体B,收率较低。文献报道了从母液中回收中间体B的非对映异构体B-S,经脱保护、消旋、结晶得到外消旋体B-rac半水合物[6],该回收工艺可提高中间体B总收率。
3 中间体C的合成路线
Boc-L-丝氨酸(2c)与2-溴-1,1-二甲氧基乙烷反应得到3c,再用甲磺酸脱保护得到片段4c;6与碘甲烷发生酯化反应得到5c,用三氟乙酸脱苄后与正己基碘反应得到6c;然后与Boc-肼反应得到7c,在碱性条件下水解得到片段8c[11]。
片段4c和片段8c在EDC介导下偶联,得到酰胺10c,用甲磺酸处理,得到11c,在碱性条件下水解得到羧酸12c,三步收率为63%。也可以用氨基酸9c与片段8c缩合,再经环合直接得到中间体12c,避免了羧基的酯化和水解,但两步收率仅为16%[11]。
文献报道了两种中间体12c脱羧制备中间体C的方法[11]。中间体12c用4,4‘-二氯二苯二硫醚和9-均三甲苯基-2,7-二甲基-10-甲基吖啶盐处理后,加入2,6-二甲基吡啶,在室温下用蓝色发光二极管照射下脱羧,经快速色谱法纯化,得到中间体C,收率92%,该方法采用常规反应设备难以放大规模,若采用连续流反应可以持续化生产。另一种为两步法,通过制备N-羟基邻苯二甲酰亚胺酯中间体13c实现,总收率为38%,该方法过程繁琐且收率较低,规模放大难度较大。
文献报道了用中间体B制备中间体C的方法[5]。苄基保护的中间体B用正己醇镁盐与1-己醇反应,然后与甲苯磺酸成盐,得到己基保护的中间体C,收率87%。
4 巴洛沙韦酯(1)的合成
4.1 按中间体A + B的方式合成
中间体A和B在1-丙基膦酸酐和甲磺酸作用下缩合,随后用溴化苄和碳酸钾处理,得到苄基保护的中间体4,收率53%;中间体4用氯化锂脱苄得到巴洛沙韦(2),收率94%,然后与3反应得到巴洛沙韦酯(1),收率93%[10]。该合成路线需要用低收率的拆分方法制备片段B,片段B和外消旋片段A偶联时非对映选择性较差,且收率较低。
4.2 按中间体A + C的方式合成
中间体A和C在1-丙基膦酸酐和甲磺酸作用下缩合,经结晶得到甲磺酸盐5,收率85%,然后5用氯化锂脱去正己基保护,得到巴洛沙韦(2),收率91%,然后与氯甲基碳酸甲酯反应得到巴洛沙韦酯(1),收率93%[11]。该合成路线使用正己基保护的片段C,其制备过程避免了低收率的拆分工艺,片段C与外消旋片段A偶联的非对映选择性较好,收率较高。

图3 巴洛沙韦酯(1)合成路线
5 结语
以路线1.5制备中间体A,条件温和,原料易得,适合工业化生产。使用正己基保护的片段C与片段A偶联的方式制备巴洛沙韦酯,非对映选择性较好,收率较高。片段C的合成路线避免了拆分工艺,收率较高且条件温和,若其光化学脱羧步骤采用连续流反应,则该路线具有更好的工业化生产前景。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
中国典型病例大全(2022年9期)2022-04-19
汕头大学学报(自然科学版)(2020年4期)2020-12-14
第二课堂(小学版)(2019年4期)2019-05-13
中成药(2017年5期)2017-06-13
股市动态分析(2015年12期)2015-09-10
小学生·多元智能大王(2015年7期)2015-07-03
中国当代医药(2015年9期)2015-03-01
中华皮肤科杂志(2014年4期)2014-12-19
中国医药科学(2013年17期)2013-09-01
