
UHPLC-MS/MS同位素内标法测定豆制品中16种喹诺酮类药物残留
2020-08-14 07:45:04廖朝选黄永桥毛敏霞王波娜王正强杨昌彪
中国调味品 2020年8期
廖朝选,黄永桥,毛敏霞,王波娜,王正强,杨昌彪
(贵州省分析测试研究院,贵阳 550014)
喹诺酮类药物(Quinolones)是近年来研究较多的一类抗菌药,因其具有抗菌谱广、高效、低毒性等优点,常作为饲料添加剂用于畜禽及水产品养殖,广泛用于动物疾病的预防和治疗以及促生长等作用[1,2]。研究发现,长期使用喹诺酮类药物会导致动物食品中存在药物残留,影响人体健康。喹诺酮类药物对中枢神经系统造成不良反应,可致重症肌无力症状加重、呼吸肌无力而危及生命,有潜在的致癌性和遗传性,且易导致病菌产生耐药性等危害[3-6],引起了人们的广泛关注。我国、日本及世界卫生组织、欧盟等国家和组织都将喹诺酮类药物列入限制使用的兽药名单中,中华人民共和国农业部2015年公布的第2292号公告中规定禁止在动物食品中使用洛美沙星、培氟沙星、氧氟沙星、诺氟沙星4种兽药。
有报道显示,为了防止豆制品变酸,延长其保质期,有商贩非法添加抗生素[7,8]。喹诺酮类主要用于动物疾病的防治,由于其优良的抗菌活性,非法用于食品生产已有相关报道[9],豆制品不易保存的特点,也是喹诺酮类抗生素潜在的非法添加目标。当前,关于食品中喹诺酮类药物的检测方法较多,多集中于动物源食品[10-12],对于水、蔬菜及麻辣烫等不同基质的检测方法也有相关报道[13-15],已经较为成熟,但关于豆制品中的检测方法研究较少。为了遏止一些不法商贩非法使用喹诺酮类抗生素,产生食品安全问题,建立豆制品中快速、准确、灵敏度高的检测方法十分必要。本研究采用同位素内标法结合UHPLC-MS/MS联用仪建立了同时测定豆制品中16种喹诺酮类药物残留的同时确证分析方法,为食品安全监管相关工作提供了可靠的技术保障。
1 材料与方法
1.1 材料与试剂
依诺沙星、氟罗沙星、诺氟沙星、氧氟沙星、培氟沙星、环丙沙星、洛美沙星、达氟沙星、恩诺沙星、奥比沙星、沙拉沙星、双氟沙星、司帕沙星、恶喹酸、萘啶酸、氟甲喹(纯度>97%)16种喹诺酮类混标溶液:100 μg/mL;诺氟沙星-D5溶液:100 μg/mL;环丙沙星-D8溶液:100 μg/mL;恩诺沙星-D5溶液:100 μg/mL,均购于天津阿尔塔科技有限公司;乙腈、甲醇(色谱纯):德国Merck公司;甲酸(色谱纯)、HLB(200 mg/6 mL):上海安谱实验科技股份有限公司;陶瓷均质子:迪马科技有限公司;实验用水为Milli-Q制备的超纯水。
1.2 仪器与设备
Agilent 1290超高效液相色谱仪、Agilent 6470 QQQ三重串联四级杆质谱(配有电喷雾离子源) 美国Agilent公司;Blixer 3搅拌机 法国ROBOT COUPA公司; LT2002电子天平 常熟市天量仪器有限公司;UMV-2多管涡旋混合器 北京普立泰科仪器有限公司;离心机 湖南湘仪离心机仪器有限公司;XW-80A涡旋混合器 上海米青科实业有限公司;Milli-Q超纯水机 美国 Millipore公司;0.22 μm PTFE针式滤膜 上海安谱实验科技股份有限公司。
1.3 方法
1.3.1 标准溶液配制
标准储备液:分别从16种喹诺酮类混标溶液、诺氟沙星-D5溶液、环丙沙星-D8溶液、恩诺沙星-D5溶液安瓿瓶中准确移取1.00 mL于不同10 mL容量瓶中,用乙腈稀释并定容至刻度,混匀,配制成浓度为10.0 μg/mL的标准储备液,-18 ℃下保存3个月。
中间标准储备液:分别准确移取0.50 mL标准储备液于不同10 mL容量瓶中,用乙腈稀释成500 ng/mL的中间标准储备液。
标准工作液:移取不同体积的中间标准储备液于不同10 mL容量瓶中,用初始流动相稀释成0.50~50.00 ng/mL的标准工作液,其中内标为5.0 ng/mL,现配现用。
1.3.2 色谱与质谱条件
色谱条件:色谱柱:ZORBAX Eclipse Plus-C18反相色谱柱(50 mm×2.1 mm,1.8 μm),美国Agilent公司;流动相A:0.1%甲酸水溶液流动相,B:乙腈;流速0.3 mL/min;柱温40 ℃;进样体积2.0 μL;梯度洗脱程序见表1。
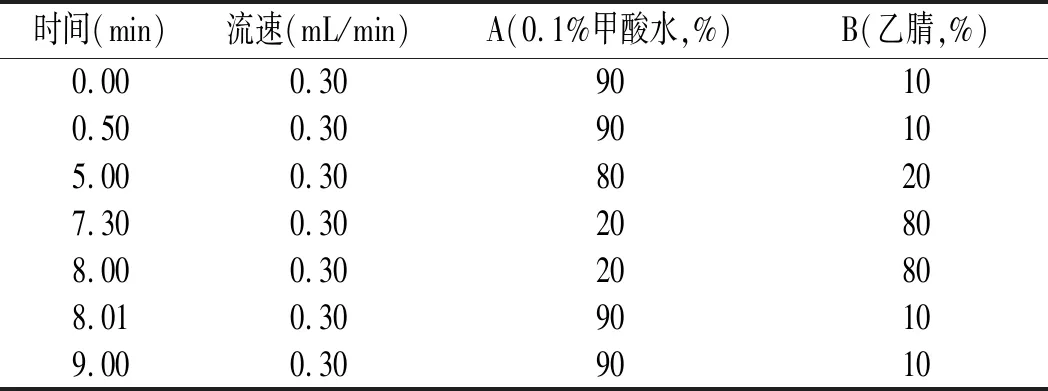
表1 流动相梯度洗脱程序Table 1 Gradient elution procedure of mobile phase
质谱条件:离子源:电喷雾离子源(ESI),正离子扫描;多反应监测(MRM);毛细管电压:4.0 kV;雾化器压力:45 psi;干燥气流速:10 L/min;干燥气温度:300 ℃;鞘气流速:10 L/min;鞘气温度:300 ℃;其他质谱条件见表2。
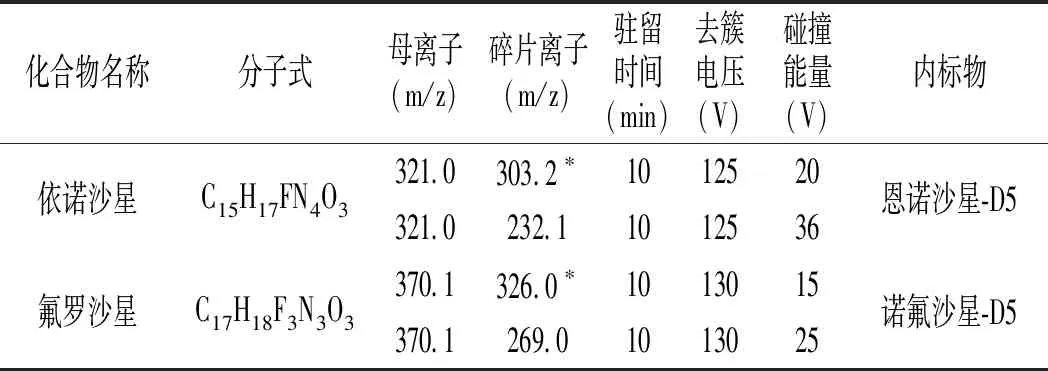
表2 质谱条件Table 2 MS parameters
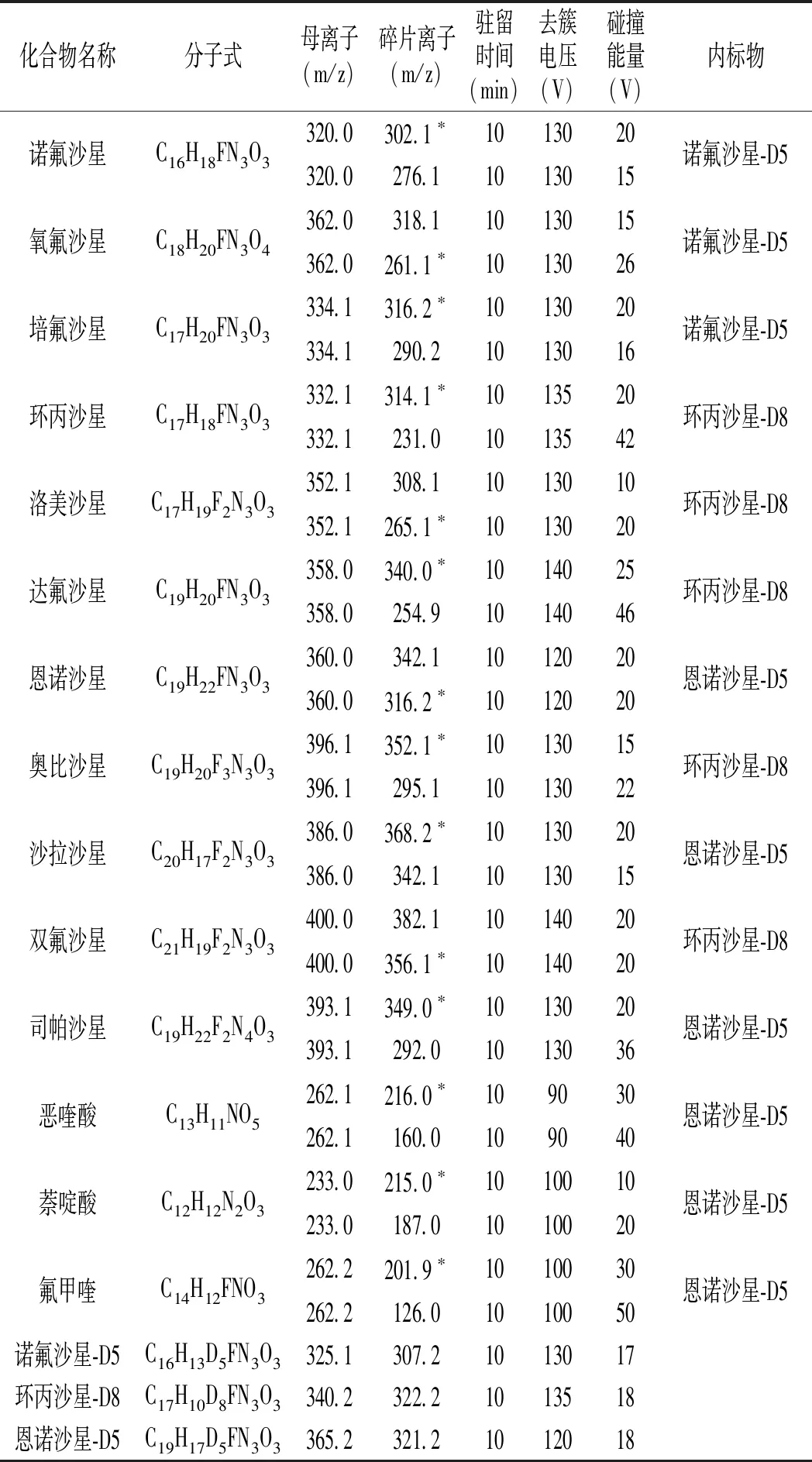
续 表
1.3.3 样品前处理
提取:称取5.0 g均匀样品(精确至0.001 g),置于50 mL离心管中,精密加入混合内标标准中间溶液0.1 mL,加入1粒陶瓷均质子,加入25 mL 0.1 mol/L EDTA-Mcllvaine缓冲溶液,涡旋混匀10 min,4500 r/min离心10 min,上清液转移至50 mL比色管中,残渣用0.1 mol/L EDTA-Mcllvaine 缓冲溶液重复提取2次,每次10 mL,合并上清液至同一比色管中,用0.1 mol/L EDTA-Mcllvaine 缓冲溶液定容至刻度,混匀。用滤纸过滤(必要时离心后过滤),续滤液待净化。
净化:精密吸取上述待净化液10.0 mL,以约1 mL/min的流速全部通过HLB固相萃取小柱(上样前依次用5 mL甲醇和5 mL水活化),用3 mL 5%甲醇水溶液淋洗,抽干,用6 mL甲醇洗脱并收集,45 ℃氮吹至近干,准确加入1.0 mL流动相初始比例复溶液溶解残渣,过0.22 μm PTFE滤膜,滤液供液相色谱-串联质谱仪分析。
2 结果与分析
2.1 前处理优化
本研究对比了使用0.1%甲酸乙腈溶液提取后,氮吹干用正己烷去除油脂和0.1 mol/L EDTA-Mcllvaine 缓冲溶液提取,经HLB固相萃取小柱净化的净化效果。结果表明,使用0.1%甲酸乙腈溶液的方法虽然快速简单,但其去除干扰物质的能力比HLB固相萃取小柱略差。实验进一步对使用涡旋振荡提取和超声提取效果及其提取时间进行了优化,最终确立样品加入0.1 mol/L EDTA-Mcllvaine 缓冲溶液,涡旋振荡提取10 min,HLB固相萃取小柱净化的样品前处理过程。
2.2 色谱条件优化
本研究采用超高效液相反相C18色谱柱对喹诺酮类化合物进行分离,因为16种喹诺酮类化合物均为正离子模式,研究表明[16]在水相中加入一定比例的甲酸能有效促进目标物的电离,故实验选用0.1%甲酸水溶液作为流动相。实验发现氟罗沙星、诺氟沙星、氧氟沙星、培氟沙星、环丙沙星等化合物很难在色谱条件下将其完全分离,实验对比了甲醇和乙腈对16种喹诺酮类药物化合物分离的影响。当使用甲醇作为有机相时,目标物虽然响应值更大,但目标物的分离度较差,尤其是氟罗沙星、诺氟沙星、氧氟沙星、培氟沙星、环丙沙星等基本重合,无法分开;使用乙腈作为有机相时,氟罗沙星、诺氟沙星、氧氟沙星、培氟沙星、环丙沙星等化合物有较好的分离度,且各化合物的响应值较好。因此综合考虑,实验使用0.1%甲酸水溶液-乙腈作为流动相体系,具体洗脱条件见表1。16种喹诺酮类化合物混标溶液的重构离子色谱图见图1。
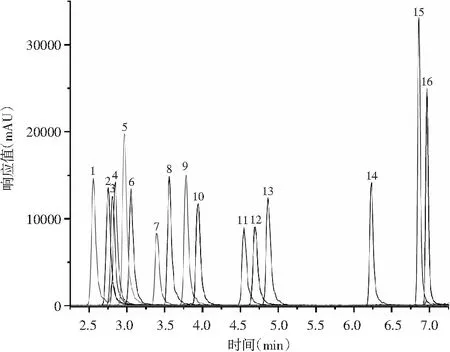
图1 16种喹诺酮类化合物混标溶液重构离子色谱图(5 μg/L)Fig.1 Reconstructed ion chromatogram of mixed standard solution of 16 quinolones注:1~16分别表示依诺沙星、氟罗沙星、诺氟沙星、氧氟沙星、培氟沙星、环丙沙星、洛美沙星、达氟沙星、恩诺沙星、奥比沙星、沙拉沙星、双氟沙星、司帕沙星、恶喹酸、萘啶酸、氟甲喹。
2.3 质谱条件选择
根据喹诺酮类药物化学结构的特点,易得到H+而形成[M+H]+准分子离子,故本研究选择正离子模式监测,采取直接接双通的方法,在正离子模式下一级全扫描质谱得到相应准分子离子峰,优化锥孔电压,再分别以准分子离子峰为母离子进行二级子离子扫描,选择特征碎片中离子中响应值高、基线噪声低、干扰小的两对离子对作为定性、定量离子对,通过优化其碰撞能量、毛细管电压、雾化器压力、干燥和鞘气的温度和压力等质谱参数,尽可能使目标物的响应值达到最大,经优化后得到表2的质谱参数。
2.4 基质效应
采用ESI源质谱仪分析食品中兽药残留时,基质效应是影响仪器灵敏度和重现性的常见因素,同位素内标法因其性质与目标物相似,在样品中损失和受基质影响程度相同,是常用来降低基质效应、提高定量准确性的一种方法[17]。实验发现,样品对16种喹诺酮类化合物均存在基质抑制效应,结果见表3,抑制效应在47.4%~90.9%之间,基质效应对目标物的影响较大。为降低基质效应,确保方法的灵敏度、重现性和定量分析的准确性,本方法在样品处理和配制标准工作液时加入已知浓度的内标物。方法选用诺氟沙星-D5、环丙沙星-D8、恩诺沙星-D5作为相应16种喹诺酮类化合物的内标物,每种化合物内标依据农业部1077号公告-1-2008[18],16种喹诺酮类化合物对应的内标物见表2。
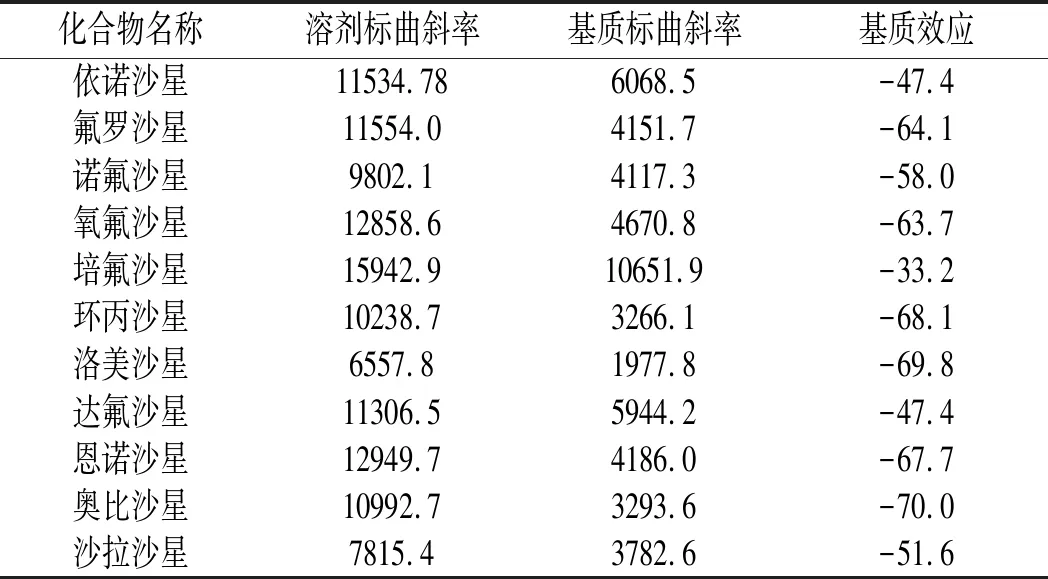
表3 16种喹诺酮类化合物基质效应Table 3 Matrix effects of 16 quinolones

续 表
2.5 方法学验证
2.5.1 标准曲线和检出限
在本研究所确定的色谱和质谱条件下,以定溶溶液配制系列质量浓度的混合标准工作溶液,绘制标准曲线,以待测物标准溶液质量浓度为横坐标,以标准品定量离子对与内标物离子对峰面积的比值为纵坐标,分别绘制16种喹诺酮类药物标准溶液工作曲线。通过向阴性样品中添加目标物来考察方法的检出限(LOD,S/N=3),见表4。结果表明,16种喹诺酮类药物在0.50~50.00 μg/L的质量浓度范围内呈现良好的线性关系,相关系数(R2)在0.99以上,检出限(LOD)均为0.50 μg/kg。
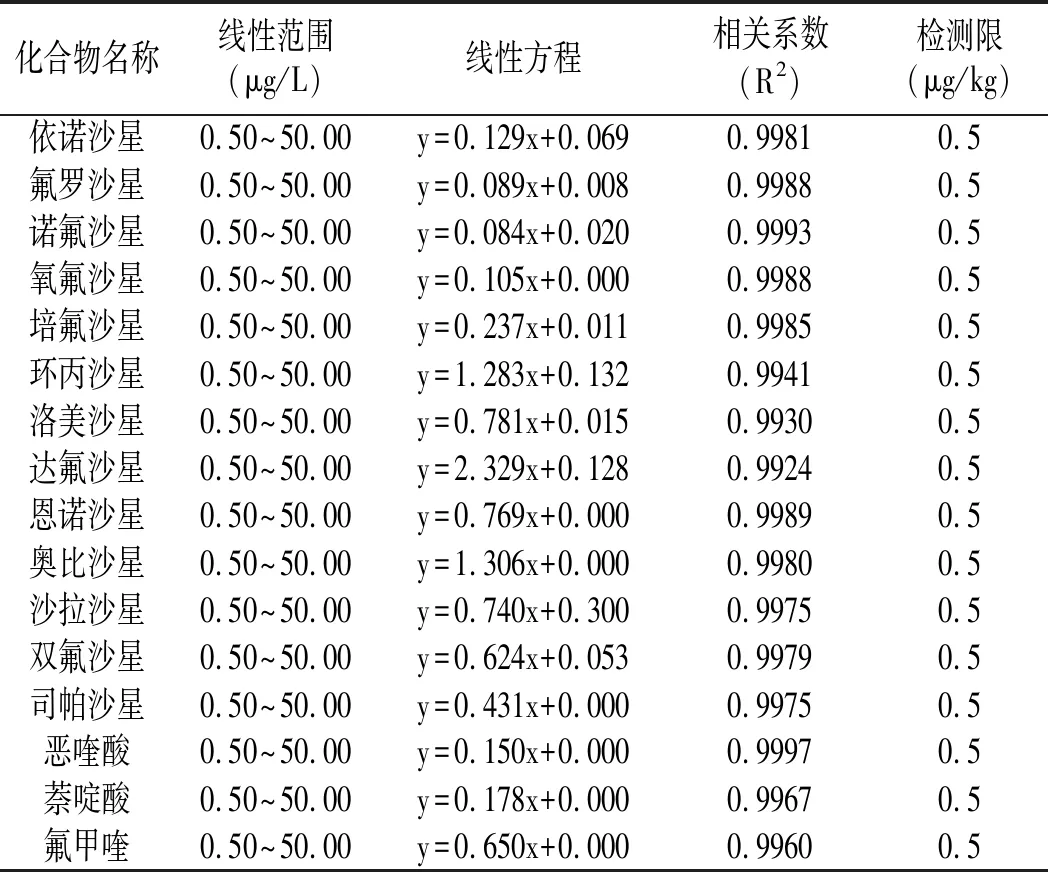
表4 线性方程、相关系数和检出限Table 4 The linear equations, regression coefficients and limits of determination
2.5.2 方法精密度和回收率
采用空白样品中添加标准溶液的方法进行加标回收和精密度实验,选取豆腐阴性样品,分别在检出限的1倍、2倍和10倍添加浓度下,每个添加水平做6个平行,计算加标回收率和相对标准偏差,豆腐样品加标色谱图见图2,结果见表5。16种喹诺酮类药物平均回收率在80.7%~107.1%之间,相对标准偏差(RSD)在1.5%~10.6%之间,说明该方法具有较高的回收率和精密度,适用于豆制品中不同浓度的喹诺酮类药物残留定性定量的检测分析要求。

图2 豆腐中16种喹诺酮类化合物加标MRM色谱图Fig.2 MRM chromatogram of 16 quinolones in tofu (5 μg/kg)
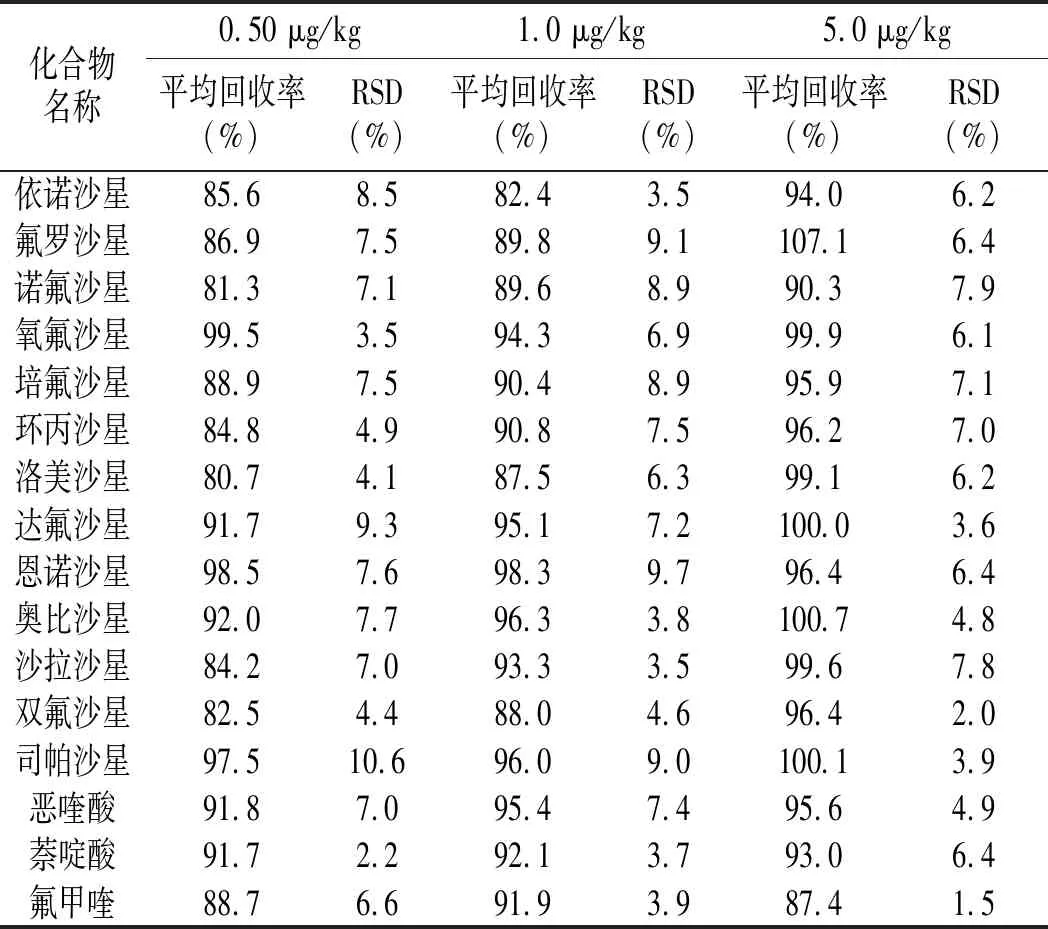
表5 加标回收率和精密度(n=6)Table 5 The recovery rates and precision (n=6)
3 结论
本研究建立了豆制品中16种喹诺酮类药物残留的同位素内标结合超高效液相色谱-串联质谱法检测分析方法。样品以0.1 mol/L EDTA-Mcllvaine 缓冲溶液提取,经HLB固相萃取小柱净化和同位素内标法补偿基质效应,使方法的灵敏度、准确度和精密度均能满足喹诺酮类药物残留分析检测的要求。实验表明,16种喹诺酮类药物在9 min内完成分析,在0.50~50.00 ng/mL质量浓度范围内,相关系数(R2)均大于0.99,检出限为0.5 μg/kg,在检出限的1倍、2倍和10倍添加浓度下,16种喹诺酮类药物平均回收率在80.7%~107.1%之间,相对标准偏差(RSD)在1.5%~10.6%之间。该方法分析结果能满足16种喹诺酮类药物对限量、回收率和精密度的分析要求,适用于豆制品中喹诺酮类药物残留的分析检测。
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02 08:23:00
昆明医科大学学报(2021年8期)2021-08-13 08:59:14
世界最新医学信息文摘(2021年12期)2021-06-09 08:36:56
保健与生活(2019年1期)2019-01-13 13:54:39
大东方(2017年10期)2017-05-30 18:29:24
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:37
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:34
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:01
兽医导刊(2016年12期)2016-05-17 03:51:52
中国卫生标准管理(2015年25期)2016-01-14 09:29:24
