
聚羟基丁酸酯合成引发的高密度生长大肠杆菌的多位点突变分析
2020-08-04 03:02陈桥吴海英王宗寿谢雨康李宜青孙俊松
生物技术通报 2020年7期
陈桥 吴海英 王宗寿 谢雨康 李宜青 孙俊松,3
(1. 中国科学院上海高等研究院,上海 201210;2. 中国科学院大学,北京 100049;3. 上海科技大学,上海 201210;4. 福建省南平市畜牧站,南平 353000)
全世界已经生产和消费了上亿吨塑料产品[1]。由于塑料垃圾不能自然降解而日积月累,对地球造成越来越大的生态危机[2]。随着公众对环境恶化和资源消耗的日益关注,近年来利用生物技术生产可降解聚合物替代制品受到了广泛关注[3]。
聚羟基脂肪酸(Polyhydroxyalkanoates,PHAs)是一些微生物在营养不均衡的条件下生长时,在胞内合成的高分子聚合物[4],作为胞内能量储存物[5]。PHAs的物理性质会因其化学组成和重复单元数量不同而各不相同[6]。聚3-羟基丁酸酯(Poly-3-hydroxybutyrate,PHB)是自然界最常见的 PHAs[7]。文献报道,将来自罗氏真养菌产PHB的操纵子phaCAB外源表达到大肠杆菌,重组大肠杆菌可以合成PHB。同样,在酿酒酵母和蜡状芽孢杆菌也实现了PHB的异源合成[8]。
罗氏真养菌中PHB的合成途径是从乙酰辅酶A开始,经phaA编码的β-酮基酯酞辅酶A硫解酶催化两个乙酰辅酶A产生乙酰乙酰辅酶A;再由phaB编码依赖于NADPH的乙酰乙酰辅酶A还原酶,催化乙酰乙酰辅酶A转化为羟基丁酰辅酶A;最后由聚羟基脂肪酸聚合酶(phaC)将羟基丁酰辅酶A单体聚合形成PHB。尽管重组大肠杆菌PHB发酵生产的水平已大幅提升,但与石油工业的衍生聚合物相比,PHB的微生物生产仍面临成本高的问题。因此,一些研究策略,如优化发酵工艺、使用农业废弃物等廉价碳源[9]及进一步改造生产菌株仍然十分必要。在菌株的改造中,除了需要敲除副产物生成的竞争途径,优化碳源代谢流及乙酰辅酶A的分配,增加还原力辅因子NADPH的供给也显著影响PHB的合成效率[10]。
此外,增加细胞密度也是提高PHB生物合成量的有效手段。研究发现,影响细胞密度的重要因素之一是发酵液中积累大量的有机酸,而其产生与pH值和培养基的组成成分相关。当培养基中碳源浓度高于生长需求时,富余碳源不被充分氧化产生二氧化碳,此部分碳代谢流会走向有机酸的合成,其中主要包括乙酸[11]。大肠杆菌在营养均衡的培养基以及微酸性发酵条件(pH 6.0-7.0)下产乙酸浓度较低,此条件下有利于细胞密度的增加。其他相关条件,尤其是碳源浓度直接影响大肠杆菌碳代谢途径的网络调控。例如,在大肠杆菌JM109中,高浓度的葡萄糖会抑制丙酮酸脱氢复合体参与其分解物丙酮酸的进一步氧化,转而利用丙酮酸氧化酶(poxB)途径脱羧生成乙酸[12]。细胞内的全局转录调控因子,如Cra、Crp和ArcA,也被证明与碳代谢和细胞密度有关[13]。
目前研究发现,各亚种的大肠杆菌菌株均可用于PHB的生产,但即使使用完全相同的表达策略,某些菌株仍表现出相对较高的PHB产量。此前我们获得了一株大肠杆菌S17-3,它是模型菌株S17-1的实验室自发突变株。在富含碳源的培养基中,表达phaCAB的S17-3转化子比其他大肠杆菌具有更好的生长性能[14]。针对大肠杆菌在PHB合成中展现的碳源利用率的提升,以及高密度培养的特异表型,十分有必要对该突变株进行深一步的研究,本研究借助全基因组测序和转录组测序分析等手段,发现了葡萄糖代谢通路中的磷酸葡萄糖异构酶(pgi)和6-磷酸葡萄糖脱氢酶(zwf)基因对于S17-3的高密度生长性能和PHB生产起着重要作用。
1 材料与方法
1.1 材料
菌株、质粒信息以及PCR 所用的模板及引物序列等信息如表1 所列,大肠杆菌S17-3为实验室保存的实验菌株;全基因组扫描测序及比对分析,上海美吉生物公司;转录组学测序及数据初步统计,生工生物工程(上海)股份有限公司;基因组DNA提取试剂盒、PCR清洁试剂盒、质粒小剂量提取试剂盒等,杭州爱思进生物技术有限公司;Taq DNA聚合酶,康为世纪生物科技有限公司;限制性内切酶BamH I 和XhoI,赛默飞世尔科技公司;Phanta高保真DNA聚合酶、一步克隆试剂盒,南京诺唯赞生物科技有限公司。S1000TMPCR扩增仪,伯乐生命医学产品(上海)有限责任公司;Tanon EPS30电泳槽及Gel DocTM XR+全自动凝胶成像仪,伯乐生命医学产品(上海)有限责任公司;NANODROP 2000c微量分光光度计,赛默飞世尔科技公司;高速冷冻离心机,艾本德生命科学仪器有限公司;恒温培养箱及恒温摇床,上海智城分析仪器制造有限公司。

表1 菌株,质粒和引物
1.2 方法
1.2.1 培养基的配制 大肠杆菌的摇瓶培养均使用LB培养基:10 g/L 氯化钠,10 g/L 胰蛋白胨,5 g/L酵母浸取物;发酵产PHB的培养基为添加了20 g/L葡萄糖,20 g/L乳糖,20 g/L甘油的LB培养基。在实验需要时,添加的氨苄青霉素(Amp)浓度为100 μg/mL。
1.2.2 质粒构建 以引物对CAB-F和CAB-R进行高保真PCR得到目标phya-CAB表达载体(引物序列见表1),将得到的PCR产物进行清洁并测定浓度。用BamH I和XhoI快切酶将质粒pBluescript II SK(+)线性化,在37℃中反应2 h后进行清洁并测定浓度。将清洁后的质粒和片段通过一步克隆试剂盒进行连接,之后立即进行转化。
1.2.3 大肠杆菌电转感受态的制备及电转方法 从大肠杆菌LB平板上挑取单克隆到3 mL的液体LB培养基中,过夜培养。按1%的接种量转接到盛有50 mL培养基的500 mL三角烧瓶里,置于37℃,200 r/min的恒温摇床培养,3-4 h待OD600到0.5-0.6左右,取出置于冰上预冷。于此同时,将电转杯,冷冻离心机,无菌水,无菌EP管也都预冷上。将预冷好的菌液放入预冷的冷冻离心机(4℃)中,5 500 r/min,7 min,去上清,留下菌体。用预冷的无菌水洗涤菌体3次后,用300 μL无菌水打匀菌体,分装到预冷的1.5 mL EP 管中。转化操作时,将2.0 μL DNA片段加入100 μL 感受态细胞中,混匀后冰浴10 min。设置电转电压为2.5 KV,空击时间为6.0 ms。电转操作后,加入700 μL 37℃的 LB培养基复苏,置于37℃恒温摇床,1 h。获得转化子后,挑取单菌落进行PCR验证,引物来自所构建质粒的同源片段[15]。
1.2.4 大肠杆菌基因敲除方法 本研究使用了Red的同源重组方法进行基因敲除。Red重组系统由3个基因编码,分别是exo,bet和gam。Exo是5'-3'核酸外切酶,可以作用于双链DNA,Bet是单链DNA结合蛋白,Gam抑制RecBCD核酸外切酶的活性。在大肠杆菌(Escherichia coli)以及酿酒酵母菌(Saccharomyces cerevisiae)中,Red重组酶可以使同源序列长只有35 bp的DNA片段与细菌的染色体发生同源重组,从而实现对细菌的定点改造。Red重组通常会在染色体基因上留下抗性标记基因,抗性标记基因的存在有可能会影响其生物学功能。利用FLP/FRT系统可以有效的解决该问题。本研究中使用的是带有Red基因的质粒pKD46以及带有flp的重组酶的质粒pCP20。为了提高敲除效率,本研究使用的同源臂为300 bp,通过overlap PCR方法得到带有同源臂及抗性筛选基因的敲除片段。
1.2.5 摇瓶发酵实验 将转入质粒的S17-3菌株转接入摇瓶进行发酵。首先在平板上挑取单克隆,转接到3 mL带有氨苄青霉素抗性的LB液体培养基中,过夜培养8-12 h,再将500 μL培养液接入装有50 mL LB的500 mL摇瓶中(1%的接种量)。将已灭菌的葡萄糖、乳糖或甘油分别加入LB培养基,使各自的终浓度达到20 g/L。置于37度摇床培养,24 h,200 r/min。
1.2.6 全基因组扫描测序及比对分析 菌株S17-3全基因组扫描测序采用的是Illumina PE测序。测序工作由上海美吉生物公司完成。对测序所得到的基因组序列进行SNP(单核苷酸多态性)和Indel(插入缺失突变)分析。BW25113在线基因组信息网址 为:https://www.ncbi.nlm.nih.gov/nucleotide/NZ_CP009273?report= genbank&log$=nuclalign&blast_rank=4&RID=4PSGBGX501R
1.2.7 转录组学测序及比对分析 总RNA的提取:按照反转录试剂盒PrimeScriptTMRT reagent Kit with gDNA Eraser说明书进行提取。随后进行转录组学测序上机前样品准备,流程:(1)rRNA去除:使用试剂盒Ribo-off rRNA Depletion Kit,具体操作步骤简写如下:a. 准备总RNA样品;b. RNA样品与探针杂交;c. RNase H消化;d. DNase I 消化;e. rRNA-depleted RNA 纯化。(2)IncRNA 文库构建:使用试剂盒VAHTSTM Stranded mRNA-seq Library Prep Kit for illumina®,具有的操作步骤简写如下:a. IncRNA片段化;b. 双链cDNA合成;c. 末端dA-Tailing;d.接头连接;e. 连接产物纯化与片段大小分选;f. 文库扩增。(3)文库质检:使用8%的PAGE胶电泳检测。(4)定量混合:为了方便等量混合便于上机检测,对回收的DNA进行精准定量,使用的是Qubit2.0 DNA 检测试剂盒。转录组学测序由上海生工生物有限公司完成,并对转录组测序结果进行基因差异表达分析。
2 结果
2.1 不同碳源条件下的大肠杆菌高密度生长
大肠杆菌在正常的摇瓶发酵条件下,其培养液最终的OD600一般约为5-10。本研究中的大肠杆菌突变菌株S17-3在含有表达质粒pBhya-CAB(S173-H)时,可以在不同碳源的LB培养基中表现出高密度生长的特性,而含有pBhya-CAB的模式菌株BW25113的转化株(BW25113-H)并未表现出同样的生长特性。作为对照,S17-3和BW25113同时表达了不含phaCAB操纵子的空载质粒pBluescript II SK(+)(S173-P)。如图 1所示,S17-3在表达pBhya-CAB且发酵培养基中加入不同碳源时,葡萄糖(Glu)作为碳源的OD600最高,可达40左右,乳糖(Lac)和甘油(Gly)作为替代碳源,其培养液的最高OD600也达到35左右。而对照菌株,含有空载质粒pBluescript II SK(+)的S17-3转化株,及其它相应的对照株,其最高OD600均低于10。

图1 大肠杆菌各菌株的生长密度的测量
2.2 全基因组扫描测序及转录组测序分析
为了揭示大肠杆菌菌株S17-3在表达质粒pBhya-CAB表现出高密度生长特性的机理。本研究将S17-3全基因组扫描测序后同BW25113比对分析,发现两株菌的遗传差异较大,它们在碳代谢、糖的转运和转录等重要功能基因上存在序列差异,如pgi、cyaA等,pgi是编码磷酸葡萄糖异构酶,催化6P-葡萄糖合成6P-果糖,参与糖酵解代谢以及参与可拉酸的其中一种单糖岩藻糖的合成;cyaA编码腺苷酸环化酶,在碳代谢调控网络中起关键作用,总结如表2所示。其中,S17-3中的基因pgi发生了点突变,722位的碱基C变成了T,造成非同义突变(碱基的改变影响氨基酸的表达),丙氨酸(GCG编码)突变成了缬氨酸(GTG编码)。另外,点突变的基因还包括调控蛋白GalB,半乳糖磷酸转移系统中的GatB;而存在移框突变的蛋白有TtdB(酒石酸脱水酶β亚基)、GalR(LacI 家族转录调控蛋白)等,但是对上述基因进行缺失操作后,并没有发现明显的遗传性状改变(数据未显)。
此外,本研究对两种培养方式下获得的细胞进行了转录组学测序及比对分析,分别是菌株S17-3/pBhya-CAB(发酵加葡萄糖)(记为S173-H),S17-3/pBluescript II SK(+)(发酵加葡萄糖)(记为S173-P),进行了转录组学测序,每个样品2个平行。测序报告及比对分析发现,上调的基因所编码的蛋白主要参与糖的转运,应激反应以及与氨基酸合成相关的代谢途径,下调部分的基因主要编码一些糖苷转移酶或转运蛋白,总结如图2所示。据文献报道,PHB的合成需要消耗大量的辅酶因子NADPH,而NADPH细胞内的主要来源之一是戊糖磷酸途径。基因zwf是磷酸戊糖途径的关键酶,编码6P-葡萄糖脱氢酶,催化6P-葡萄糖合成6P-葡萄酸内脂,因此考察Zwf的表达量可以用来预测PPP途径的代谢流量。
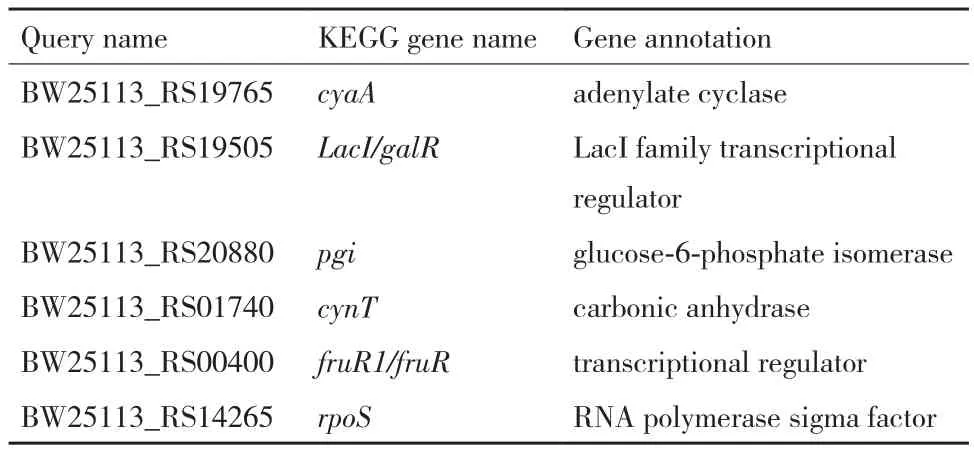
表2 全基因组扫描测序及比对分析
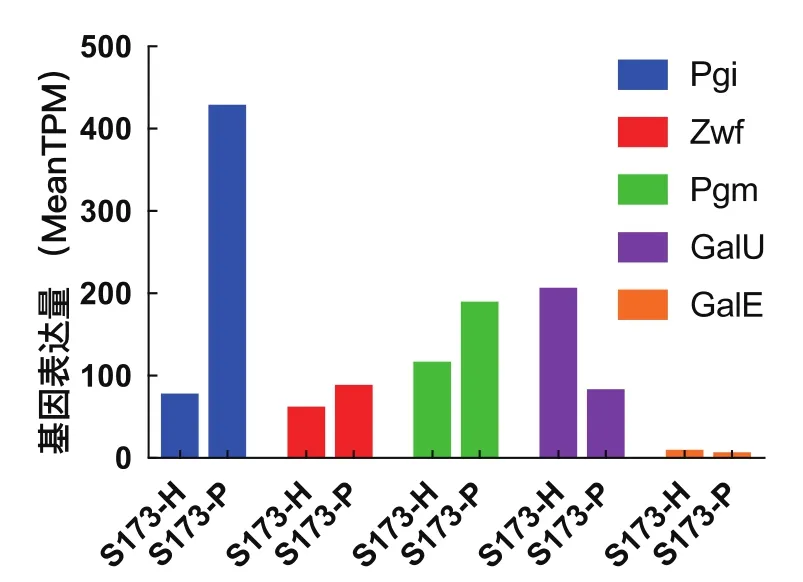
图2 转录组测序分析
2.3 突变菌株的生长曲线
为了研究基因Pgi突变带来的影响,在菌株S17-3和BW25113中,利用Red同源重组系统敲除基因pgi,得到突变菌株S17-3/Δpgi和BW25113/Δpgi。在相同的培养条件下发酵菌株BW25113,S17-3,BW25113/Δpgi和 S17-3/Δpgi。实验结果如图 3-A所示,S17-3敲除Pgi的菌株生长变得迟缓,但其最大OD600值和野生型没有显著差异。BW25113敲除Pgi的菌株生长得到改善,其最大OD600值(约9.0)也优于野生型(约5.0)。同样,在上述的培养条件下发酵菌株 S17-3/Δpgi/pBhya-CAB(S173/Δpgi-H),S17-3/pBhya-CAB(S173-H),BW25113/Δpgi/pBhya-CAB(BW25113/Δpgi-H)以及 BW25113/pBhya-CAB(BW25113-H)。实验结果如图3-B所示,S173/Δpgi-H相比S173-H同样生长变得迟缓,但其最大OD600值没有显著差异。而BW25113/Δpgi-H相比于BW25113-H的生长性能有明显提升,最大OD600值为15左右。
为了研究Zwf对S17-3高密度生长的影响,在S17-3和BW25113中分别敲除zwf,得到突变菌株S17-3/Δzwf和 BW25113/Δzwf。在上述的培养条件下发 酵 菌 株 S17-3/Δzwf,BW25113/Δzwf,S17-3/Δzwf/pBhya-CAB(S173/Δzwf-H) 和 BW25113/Δzwf/pBhya-CAB(S173/Δzwf-H)。 实 验 结 果 如 图 3-C 所 示,S173/Δzwf-H的生长受到影响,最大OD600值约为20.0,其他敲除zwf的菌株最大OD600值与野生型无显著差异。
3 讨论
随着地球资源日益减少,利用微生物发酵技术替代化学资源生产大宗化学品及可降解材料是研究的热门和重点之一。微生物的高密度生长对发酵产物的提高往往至关重要,而微生物通常会通过其复杂的代谢调控网络对细胞生长密度进行微妙而精确的调控,在不进行发酵干预的情况下,其最终培养密度往往不是很高。在本研究中,我们通过在一株大肠杆菌突变菌株S17-3中表达质粒pBhya-CAB,获得了高密度生长的重组大肠杆菌。借助全基因组测序和转录组测序和比对分析,发现pgi有基因突变和转录水平的改变,zwf则在转录水平上有改变。在S17-3和BW25113中分别敲除pgi和zwf,结果显示S17-3敲除pgi后,菌株生长会变得缓慢,但最大OD600值无显著差异;BW25113敲除pgi后,菌株的生长性能得到改善,表达pBhya-CAB时最大OD600值较之前提升了约3倍。S17-3敲除zwf后,菌株生长性能受到影响,表达pBhya-CAB时最大OD600值降低了约1倍。BW25113敲除zwf后,菌株生长变缓慢,但最大OD600值无明显差异。
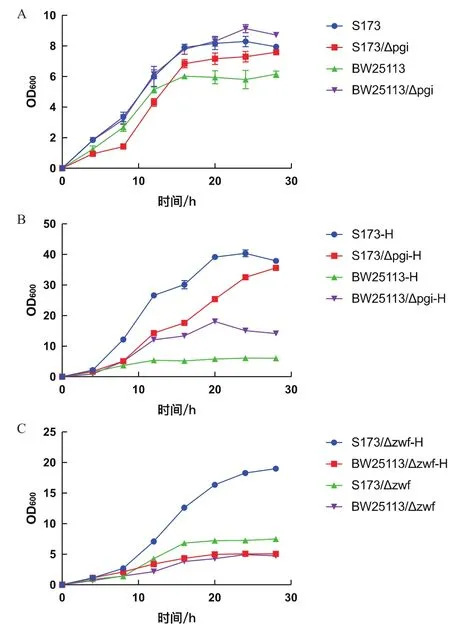
图3 大肠杆菌敲除Pgi和Zwf的生长曲线
研究发现,如图4的代谢示意图所示,pgi的敲除能提高菌体内的磷酸戊糖途径(Pentose phosphate pathway),导致细胞内的辅酶NADPH浓度上升,而在PHB的生物合成过程中,PhaB催化需要消耗大量辅酶NADPH,从而可以提升乙酰CoA合成PHB的碳代谢支流,解释了研究中发现的敲除pgi且同时表达PHB基因时,菌株为何能高密度生长,同时高水平表达PHB的生理现象[16]。此外,之前的研究发现,该转化株产乙酸水平低,也可能是由于乙酰CoA积累变少造成的。同时,本研究发现,基因zwf的敲除不利于大肠杆菌转化株在高碳源培养基中的高密度生长(相比S17-3-H),这应该是由于,该突变会阻断磷酸戊糖途径,降低细胞内的辅酶NADPH[17],形成与pgi突变相反的代谢效果(代谢路径如图4所示)。
4 结论
通过在大肠杆菌中表达质粒pBhya-CAB,本研究获得了一株高密度生长的重组大肠杆菌菌株,无发酵调控情况下,培养液的最大OD600值可达到40左右。借助全基因组测序和转录组测序和比对分析,发现菌株S17-3的pgi有基因突变和转录水平的改变,zwf有转录水平的改变。在S17-3和BW25113中分别敲除pgi和zwf后,大肠杆菌在发酵过程中呈现出细胞密度的改变,提示S17-3的碳代谢流的分配调控网络与其他大肠杆菌菌株有明显不同,需要进一步深入的研究。
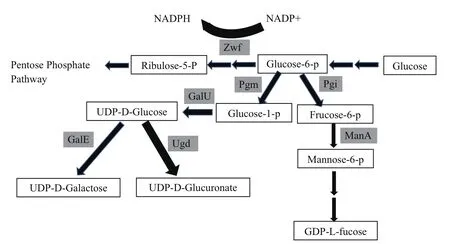
图4 本研究中所涉及大肠杆菌的相关碳代谢流走向图
猜你喜欢
分子催化(2022年1期)2022-11-02
中老年保健(2022年4期)2022-08-22
环境工程技术学报(2022年3期)2022-06-05
中国土壤与肥料(2021年5期)2021-12-02
昆钢科技(2021年6期)2021-03-09
天津科技(2020年4期)2020-05-09
中外医疗(2016年15期)2016-12-01
中外医疗(2015年11期)2016-01-04
云南中医学院学报(2015年2期)2015-07-31
质谱学报(2015年5期)2015-03-01
