
原位硅烷化改性对Au/TS-1 催化丙烯氢氧环氧化制环氧丙烷反应性能的影响
2020-07-13 09:58:16张志华段学志周兴贵
化学反应工程与工艺 2020年6期
张志华,王 刚,杜 威,段学志,周兴贵
华东理工大学化学工程联合国家重点实验室,上海 200237
自1998 年发现Au/TiO2催化剂可以用来催化丙烯与H2/O2直接反应生成环氧丙烷(PO)以来[1-2],丙烯氢氧环氧化制PO 这一新工艺因其绿色环保、产物易分离和操作简单等优势而受到工业界和催化界的广泛关注。目前,国内外的学者围绕含钛材料(如Ti-MCM-41,Ti-MCM-48,Ti-TUD,Ti-HMS和TS-1 等)性质对催化剂性能的影响展开了大量研究[3-10]。其中,分子筛TS-1 因为富含孤立的Ti4+物种,将纳米金颗粒固载在TS-1 上制备的Au/TS-1 催化剂显示出较优的活性。
研究发现,Au/TS-1 催化剂通常在20 h 内PO 生成速率下降约30%,由此制约了丙烯氢氧环氧化制氧丙烷反应(HOPO)工艺的工业化应用[8-11]。Feng 等[12]研究发现,焦炭堵塞TS-1 微孔孔道是Au/TS-1催化剂失活的主要原因。基于该失活机理,将纳米金颗粒选择性沉积在未焙烧TS-1-B 外表面上制备得到的Au/TS-1-B 催化剂具有更高的稳定性[12]。值得注意的是,Au/TS-1-B 催化剂外表面吸附的模板剂分子会覆盖部分Ti 活性位点[13-15]。采用H2/N2气氛高温还原处理的Au/TS-1-B 催化剂,虽然会降低外表面吸附模板剂的含量,提高了暴露的Ti 活性位点数量,但同时也会导致更多的Si—OH 暴露出来[14],降低了催化剂表面疏水性,导致在Ti—OH 活性位点上生成的PO 与相邻Si—OH 作用生成难脱附的碳沉积物种,从而抑制了丙烯分子在Ti—OH 活性位点上的吸附[16-17]。此外,由于纳米金颗粒的塔曼温度较低(约为300 ℃),在高温还原Au/TS-1-B 催化剂的过程中纳米金颗粒会发生团聚[18-20]。因此,开发一种在催化剂高温还原过程中既能降低表面Si—OH 数量又能抑制纳米金颗粒团聚的修饰方法,成为进一步调控Au/TS-1-B 催化剂性能亟需解决的问题。
Sinha 等[5]通入硅烷对高温还原后的Au/Ti-TUD-1 催化剂进行硅烷化改性后,显著降低了催化剂表面硅羟基数量。这有利于促进亲水性PO 在催化剂表面的脱附,又增加了丙烯分子在催化剂表面的覆盖度,因此,硅烷化改性后的Au/Ti-TUD-1 催化剂在HOPO 反应中表现出优异的性能。Chowdhury 等[16]发现,在硅烷化改性后的Au/Ti-TUD-1 催化剂反应过程中,通入三甲胺后能够同时对纳米金颗粒和载体表面起到修饰作用,进一步提高了该催化剂的性能。但高温还原催化剂后再通入硅烷及三甲胺进行改性的方式,在高温活化催化剂过程中无法起到保护纳米金颗粒的作用,而且三甲胺有毒、易燃易爆,并具有腐蚀性。因此,在催化剂高温还原过程中通入不含氮的硅烷进行原位改性处理,是一种能够降低催化剂表面Si—OH 数量并同时能够保护纳米金颗粒且安全环保、简单易行的改性新策略。
本工作将在Au/TS-1-B 催化剂高温还原过程中通入六甲基二硅氧烷(HMDSO),研究原位硅烷化改性处理来调控催化剂结构及性能,考察原位硅烷化改性Au/TS-1-B 催化剂与未改性Au/TS-1-B 参比催化剂在HOPO 反应中的性能,并结合傅里叶变换红外(FT-IR)、高角度环形暗场-扫描透射电子显微镜(HAADF-STEM)、紫外/可见/近红外光谱(UV/Vis/NIR)及X 射线光电子能谱(XPS)等表征方法分析原位硅烷化改性过程对Au/TS-1-B 催化剂上的Au-Ti-Si 位点结构的影响,探讨原位硅烷化改性对Au/TS-1-B 催化剂性能的调控机制。
1 实验部分
1.1 催化剂制备
参照文献方法[21-22]进行催化剂的制备。以正硅酸四乙酯为硅源,钛酸四丁酯为钛源,四丙基氢氧化铵为模板剂,按一定物质的量之比配料。搅拌均匀后装入带有聚四氟乙烯内衬的不锈钢水热釜中,然后将水热釜置于恒温烘箱(165 ℃)中晶化。晶化结束后,从烘箱中取出水热釜,降至室温后将浆液离心后干燥,得到未焙烧钛硅分子筛TS-1-B。以尿素为沉淀剂,采用沉积-沉淀法制备Au/TS-1-B催化剂。采用等离子体-原子发射光谱(ICP)测得所制备的Au/TS-1-B 催化剂上金的负载量为0.08%(质量分数)。其中,原位硅烷化改性催化剂命名为Au/TS-1-B(in-situ silylated),未改性催化剂命名为Au/TS-1-B(unsilylated)。
1.2 催化剂表征
金的负载量测定在等离子体发射光谱仪(Agilent 725 ICP-OES 型,美国)上进行,测试前将催化剂溶于王水与氢氟酸组成的混合溶液中,得到含有一定浓度的金溶液后再进行分析。Au/TS-1-B 催化剂上硅羟基、骨架钛及物理吸附水分子的特征峰分析采用傅里叶-红外光谱仪(FT-IR,Nicolet 6700型,美国)分析,分辨率2.0 cm-1,KBr 压片法。采用高角环形暗场像扫描透射电镜(HAADF-STEM,Tecnai G2 F20 S-Twin 型,荷兰)分析催化剂上纳米金颗粒的粒径分布。测试条件为:最大加速电压200 kV,晶格分辨率0.102 nm,点分辨率0.24 nm,设置能谱能量分辨率为136 eV,放大倍数110 万倍。拍摄前先将样品在乙醇溶液进行超声5~10 min 后再滴加约1~2 滴上层悬浊液于超薄碳膜上,然后将样品在室温下静置至乙醇挥发完后再进行测试。采用Nano Measurer 1.2 软件对Au/TS-1-B 催化剂上纳米金颗粒粒径分布进行统计分析。采用紫外/可见/近红外漫反射仪(UV/Vis/NIR,日本UV-3600 plus 型与美国Nicolet IS5 型)上分析催化剂上纳米金颗粒的表面等离子体共振峰,将样品与BaSO4混合均匀后再压片进行测试。采用X 射线光电子能谱(XPS)分析催化剂上金与钛物种的价态及组成。样品分析条件为:高压14.0 kV,通能93.9 eV,功率300 W,铝靶,先采集样品的0~1 200 eV 全扫描谱,然后采集金与钛的窄扫描谱,以C1s 为284.6 eV 作为基准对结合能进行校正,数据的分峰与拟合在XPS Peak4.1 软件上进行。
1.3 催化剂性能评价
催化剂活化处理及评价装置如图1 所示。常压固定床反应器由石英管、不锈钢壳层、保温层、加热设备以及中空铜管组成,其中内径约为0.6 mm 的石英管置于中空铜管中。装有热电偶的玻璃套管从反应器底部插入石英管中测量催化剂床层温度,在热电偶玻璃套管的顶端装填石英棉,筛分后的催化剂装填在石英棉上。原料气在混合器中混合均匀后从上往下进入反应器,其流量由质量流量计控制,从反应器出口流出的物料进入气相色谱仪进行在线分析。
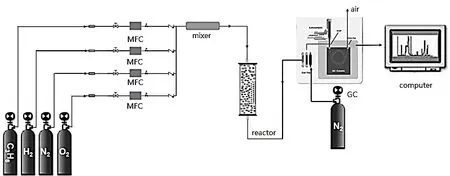
图1 催化剂评价装置示意图Fig.1 The experimental apparatus for propene epoxidation with H2 and O2
Au/TS-1-B 催化剂的原位硅烷化改性过程如下:称取0.15 g 催化剂(过100 目筛子筛分)置于石英管中,再通入H2,N2及HMDSO/N2混合气(H2和N2体积比为1:2,气体总流量为52 mL/min),并以1 ℃/min 的升温速率将催化剂柱从室温加热到300 ℃,然后活化处理1.5 h。未改性Au/TS-1-B催化剂的活化不需要通入HMDSO/N2混合气,其余条件与Au/TS-1-B 催化剂的原位硅烷化改性过程相同。
催化剂评价按如下方式进行:待反应器降温至200 ℃后,通入原料气(C3H6,H2,O2和N2的体积比为1:1:1:7,35 mL/min)进行反应,反应器出口的物料通入气相色谱仪(GC 2060,上海锐敏)中进行分析,色谱载气为高纯氮气。乙醛、环氧丙烷、丙烯醛、丙醛和丙酮的分析采用Rt-QS-BOND毛细管色谱柱(0.5 mm×30 m),氢火焰检测器(FID),柱温100 ℃,柱压70 kPa;氢气、氧气和二氧化碳的分析采用TDX-01 色谱柱(3 mm×3 m),热导检测器(TCD),柱温100 ℃,柱压40 kPa;
通过归一化法计算转化率(X)、产物的生成速率(r)和选择性(S),计算公式如下所示:

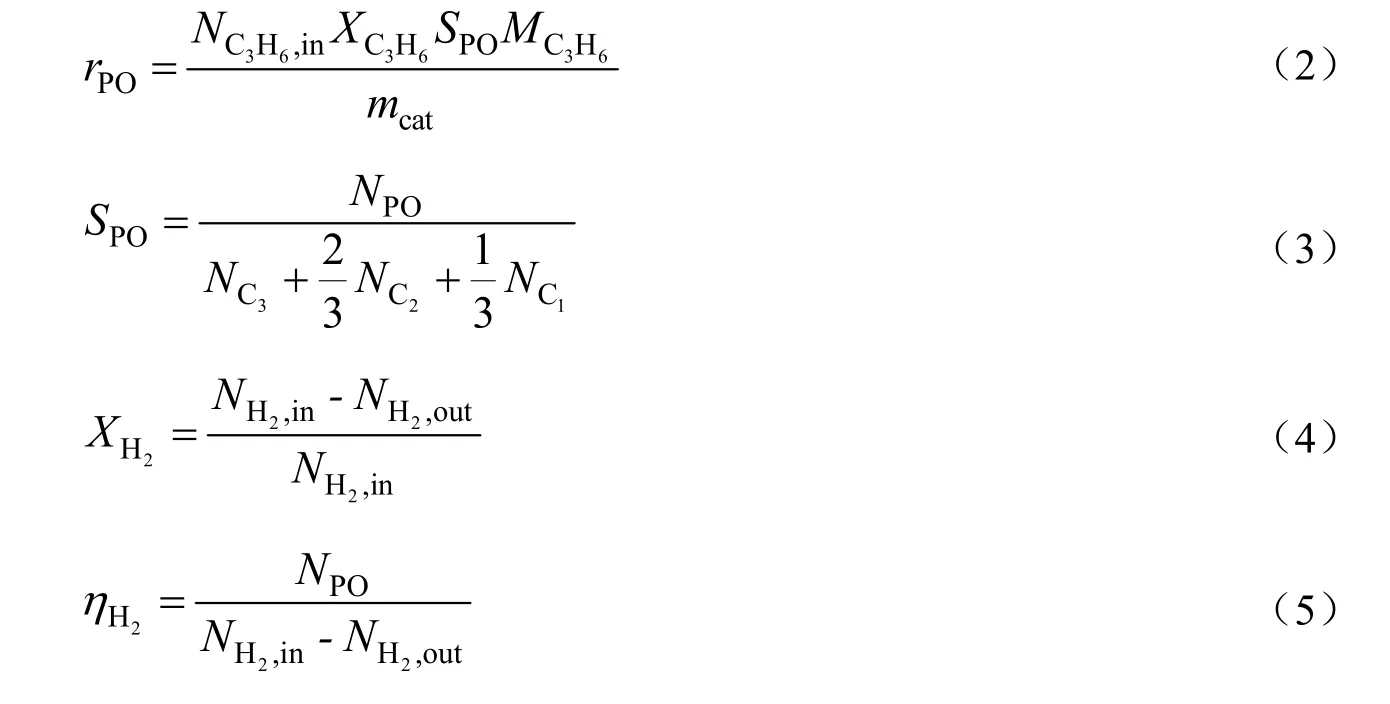
其中,N为摩尔流量,mol/h;M为分子量,g/mol;mcat为催化剂质量,kg;η为氢效。
2 结果与讨论
2.1 催化剂表征分析
FT-IR 可用于分析钛硅分子筛的骨架构型和判断Ti 是否进入分子筛的骨架结构中。原位硅烷化改性Au/TS-1-B 催化剂与未改性Au/TS-1-B 催化剂的FT-IR 谱图(图2)显示,两个催化剂均在550 cm-1处出现归属于MFI 型分子筛的特征峰,这说明硅烷化改性过程未对TS-1-B 分子筛的拓扑结构造成破坏[12-13,23-25]。从图2 还可以看出,两种催化剂在960 cm-1处出现归属为Ti 进入TS-1 分子筛骨架后形成Si—O—Ti 键的伸缩振动特征峰,并且这两个催化剂在960 cm-1处的吸收峰强度没有明显的差异,这说明硅烷化改性过程也未对TS-1-B 分子筛中的Ti 物种类型与含量产生影响[12-13,24-25]。
FT-IR 也被用来分析钛硅分子筛表面羟基及吸附水分子的特征峰。钛硅分子筛在约1 630 cm-1处出现的吸收峰通常被归属为Si—OH 特征峰,而在约3 460 cm-1处出现的吸收峰被归属为与Si—OH特征峰及吸附在Si—OH 上水分子的特征峰[4,15,26-27]。由图2 可知,相比于未改性Au/TS-1-B 催化剂,原位硅烷化改性的Au/TS-1-B 催化剂在约为1 630 cm-1和3 460 cm-1处的特征峰强度大大减弱,并且在2 800~3 000 cm-1出现的归属为C—H 键伸缩振动特征峰的强度却明显提高,这说明原位硅烷化改性过程中硅烷分子成功嫁接到了催化剂表面,从而有效降低了催化剂表面Si—OH 的数量,提升了催化剂的表面疏水性[5,15,26,28-29]。
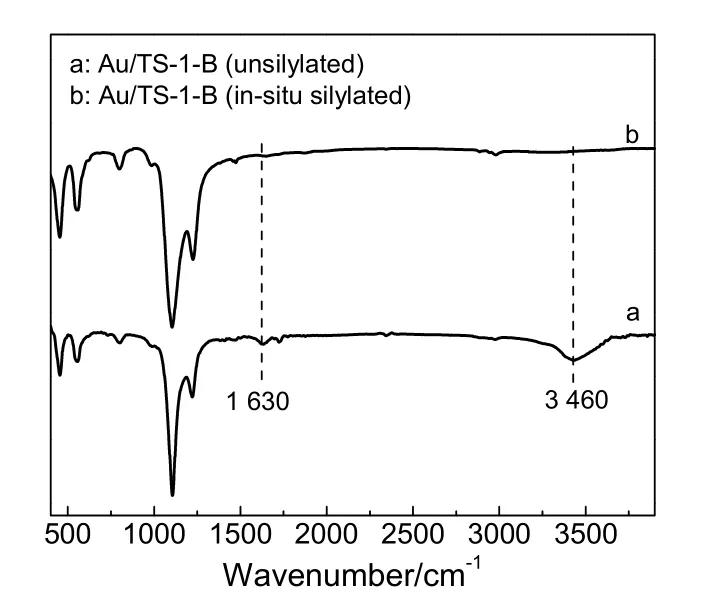
图2 Au/TS-1-B 催化剂的FT-IR 图谱Fig.2 FT-IR spectra of Au/TS-1-B catalysts
图3 为原位硅烷化改性Au/TS-1-B 催化剂与未改性Au/TS-1-B 催化剂的HAADF-STEM 图以及Au 颗粒粒径分布。从图3 可以看出,相比于未改性Au/TS-1-B 催化剂,原位硅烷化改性后Au/TS-1-B催化剂的纳米金颗粒平均粒径均有所降低,且尺寸较小的纳米金颗粒数量增多。其原因可能是在还原过程中通入HMDSO 时催化剂表面成功嫁接上了硅烷分子,这就像是在催化剂表面形成了许多“分子围栏”,有利于抑制纳米金颗粒在高温还原过程中发生团聚[16,28]。
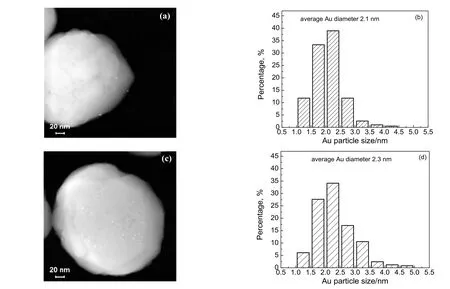
图3 Au/TS-1-B 催化剂的HAADF-STEM 图及对应的Au 颗粒尺寸分布Fig.3 HAADF-STEM images of Au/TS-1-B catalysts and corresponding Au particle size distributions(a), (b): in-situ silylated;(c), (d): unsilylated Au/TS-1-B catalysts
图4 为原位硅烷化改性Au/TS-1-B 催化剂与未改性Au/TS-1-B 催化剂的UV/Vis/NIR 图。由图4 可以看出,原位硅烷化改性 Au/TS-1-B 与未改性Au/TS-1-B 催化剂均在13 000~25 000 cm-1处出现纳米金颗粒的表面等离子体共振峰[16],并且原位硅烷化改性后Au/TS-1-B 催化剂上纳米金颗粒的表面等离子体共振峰出现了蓝移,表明纳米金颗粒的表面介电常数发生了改变[16,30]。这可能是由于部分HMDSO 在高温下裂解形成的小分子硅化合物(如硅醇)吸附在纳米金颗粒表面所致,也可能是原位硅烷化改性后Au/TS-1-B 催化剂上纳米金颗粒粒径较小的另一个原因。上述FT-IR 与UV/Vis/NIR 结果表明,在催化剂还原过程中通入HMDSO 进行原位硅烷化改性不但对载体表面进行了改性,而且也对纳米金颗粒表面进行了修饰。
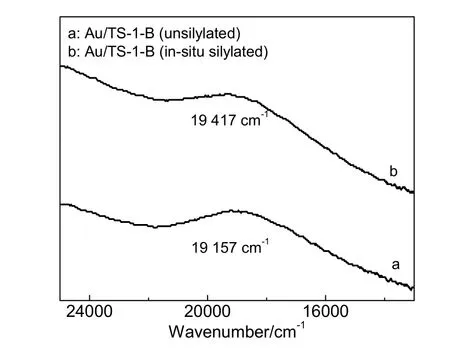
图4 Au/TS-1-B 催化剂的UV/Vis/NIR 图谱Fig.4 UV/Vis/NIR spectra of Au/TS-1-B catalysts
图5 为原位硅烷化改性Au/TS-1-B 催化剂与未改性Au/TS-1-B 催化剂的XPS 图。从图5 可以看出,原位硅烷化改性后的Au/TS-1-B 催化剂上Au0的结合能要明显高于未改性Au/TS-1-B 催化剂。这可能是由于纳米金颗粒表面吸附了少量由HMDSO 分解形成的硅醇,由于硅醇分子中的氧原子具有较强的电负性,其存在的吸电子效应可能会从金颗粒表面拿走电子而使该催化剂上Au0的结合能偏高。此外,根据HADDF-STEM结果推测,原位硅烷化改性后Au/TS-1-B催化剂上可能存在较多的尺寸较小的金物种,这部分金物种的价态相比于纳米金颗粒更偏向于氧化态,这也会使得该催化剂上Au0的结合能偏高[31]。图5 的XPS 结果显示,原位硅烷化改性Au/TS-1-B 催化剂与未改性Au/TS-1-B 催化剂的Ti 物种结合能不存在明显差异,这与Chowdhury 等[32]的研究结果一致,原因可能是由于硅烷化改性过程中硅烷分子优先与Si—OH 发生作用,因此对Au/TS-1-B 催化剂上Ti 物种的影响较小。
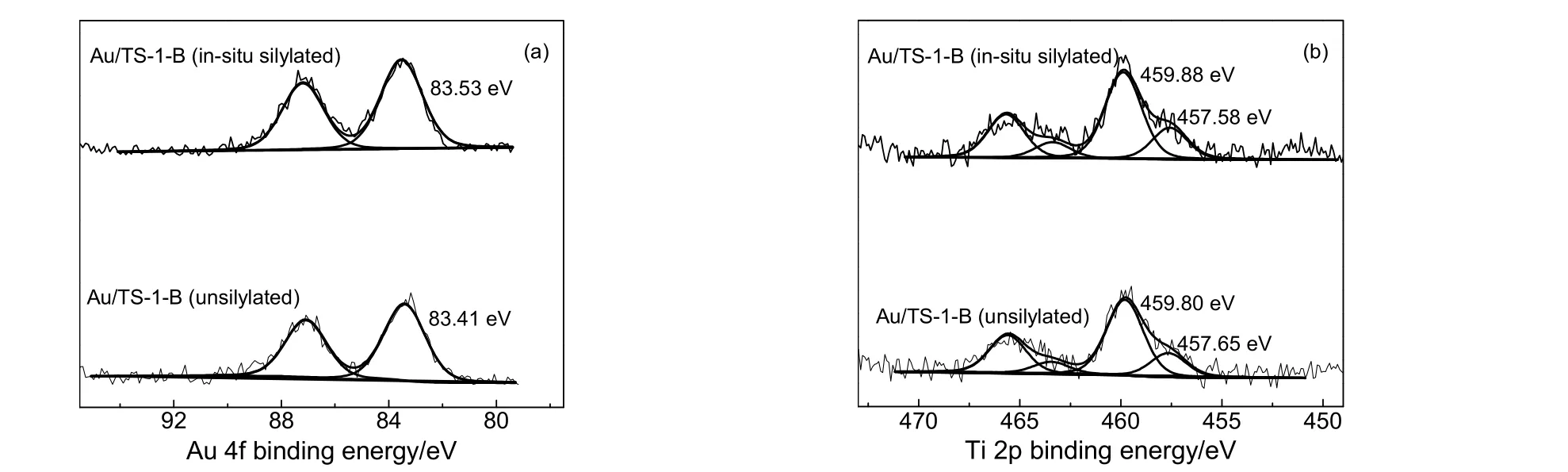
图5 Au/TS-1-B 催化剂的XPS 图谱Fig.5 XPS spectra of Au/TS-1-B catalysts
2.2 催化剂性能评价
原位硅烷化改性Au/TS-1-B 与未改性Au/TS-1-B 催化剂上丙烯转化率与PO 生成速率随时间的变化关系如图6 所示。从图6(a)中可以看出,原位硅烷化改性Au/TS-1-B 催化剂上丙烯转化率(约5.9%)明显高于未改性的催化剂(约4.5%)。此外,原位硅烷化改性Au/TS-1-B 催化剂上PO 生成速率高达203 gPO/(h·kgcat),约为未改性Au/TS-1-B 催化剂上PO 生成速率[约155 gPO/(h·kgcat)]的1.3 倍。上述结果表明,对Au/TS-1-B 催化剂进行原位硅烷化改性处理可以大幅提高丙烯转化率及PO 生成速率,这说明对Au/TS-1-B 催化剂进行原位硅烷化改性是提高催化剂性能行之有效的策略。
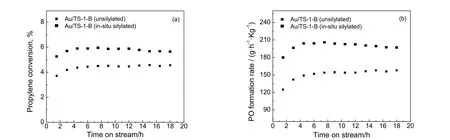
图6 Au/TS-1-B 催化剂催化丙烯氢氧环氧化反应结果Fig.6 Results of propylene epoxidation with H2 and O2 over Au/TS-1-B catalysts
原位硅烷化改性Au/TS-1-B 与未改性Au/TS-1-B 催化剂上PO 选择性和氢气转化率及氢效结果如表1 所示。由表1 可知,原位硅烷化改性Au/TS-1-B 催化剂的PO 选择性略微高于未改性Au/TS-1-B催化剂。因为PO 分子吸附在催化剂表面羟基位点上会发生开环、异构反应生成副产物[16-17],原位硅烷化改性处理显著降低了Au/TS-1-B 催化剂表面硅羟基的含量,这有利于加速PO 分子在催化剂表面的脱附,使得原位硅烷化改性Au/TS-1-B 催化剂的丙烯转化率远高于未改性Au/TS-1-B 催化剂的情况下,其PO 选择性仍略高于后者。
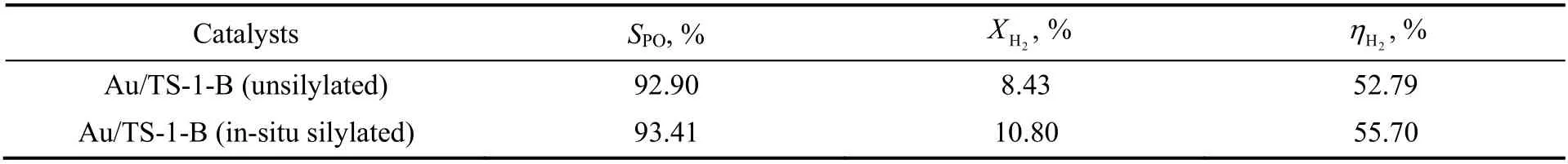
表1 丙烯氢氧环氧化反应的PO 选择性和氢气转化率及氢效Table 1 PO selectivity and hydrogen conversion as well as hydrogen efficiency in propylene epoxidation with H2 and O2
由表1 还可知,相较于未改性Au/TS-1-B 催化剂,原位硅烷化改性Au/TS-1-B 催化剂上氢气转化率明显提高,这说明该催化剂上有更多的氢气分子参与了纳米金颗粒上原位生成HOOH 物种的反应,也意味着该催化剂上HOOH 物种生成速率较高。先前的研究发现,HOOH 物种在向相邻Ti4+活性位点传递过程中会发生分解,导致氢气有效利用效率(即氢效)降低[33]。然而催化剂评价结果(见表1)显示,原位硅烷化改性Au/TS-1-B 催化剂相较于未改性Au/TS-1-B 催化剂,其对氢气转化率明显提高,而此条件下其氢效仍略高于未改性Au/TS-1-B 催化剂的值,表明原位硅烷化改性过程也有利于提高Au/TS-1-B 催化剂的氢效。
2.3 催化剂结构-性能关系分析
根据催化剂结构表征结果推测:原位硅烷化改性处理Au/TS-1-B 催化剂时,表面嫁接的硅烷分子在催化剂表面形成了“分子围栏”,起到了抑制高温还原过程中纳米金颗粒发生团聚的作用,使得原位硅烷化改性Au/TS-1-B 催化剂上纳米金颗粒平均粒径明显小于未改性催化剂上纳米金颗粒平均粒径,促使原位硅烷化改性的催化剂暴露出了更多的Au 位点,从而有利于促进更多的氢气分子参与反应,最终提高了HOOH 物种的生成速率[23,31,34]。此外,UV/Vis/NIR 结果显示,原位硅烷化改性后Au/TS-1-B 催化剂上纳米金颗粒的表面等离子体共振峰出现蓝移,这预示着该催化剂上的纳米金颗粒表面可能吸附有硅烷分解形成的硅醇分子,使得该催化剂上零价金的结合能明显高于未改性Au/TS-1-B 催化剂上零价金的结合能,有利于增强Au―H 之间的相互作用,从而有助于促进氢气分子在纳米金颗粒表面发生解离生成HOOH 物种[14,35-37]。因此,原位硅烷化改性对Au 活性位点的修饰促进了催化剂表面HOOH 物种的生成,使原位硅烷化改性Au/TS-1-B 催化剂相比于未改性催化剂,其丙烯转化率和PO 生成速率显著提高。
原位硅烷化改性也会对Au/TS-1-B 催化剂表面的Ti 位点和Si 位点产生影响。研究发现[38],HOPO反应过程中产生的H2O 和PO 分子会以单分子层的形式吸附在催化剂表面的羟基位点上,这会抑制丙烯在催化剂表面的吸附。因此,原位硅烷化改性Au/TS-1-B 催化剂表面的Si―OH 含量明显降低,这有助于促进反应过程中生成的H2O 分子在催化剂表面的脱附,也有利于抑制在Ti―OH 位点上生成的PO 吸附在相邻的Si—OH 上,加快了PO 在Ti—OH 位点上发生脱附,提高催化剂表面单位时间内暴露的Ti 活性位点数量,从而促进了丙烯分子在催化剂表面的吸附,提高了丙烯分子在催化剂表面的覆盖度。这可能是原位硅烷化改性Au/TS-1-B 催化剂相比于未改性催化剂,其丙烯转化率和PO 生成速率显著提高的另一个原因。
根据原位硅烷化改性Au/TS-1-B 催化剂与未改性催化剂性能的差异并结合催化剂结构表征,得到如图7 所示的原位硅烷化改性对Au/TS-1-B 催化剂修饰改性的机理。原位硅烷化改性能够同时对Au/TS-1-B 催化剂上的Au,Ti 及Si 位点进行修饰改性,从而提高了Au/TS-1-B 催化剂的性能。
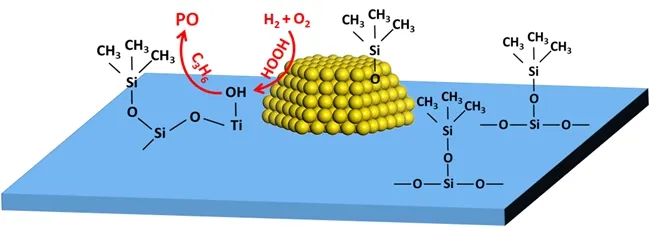
图7 原位硅烷化改性Au/TS-1-B 催化剂上PO 生成过程示意图Fig.7 Probable pathways for PO synthesis over in-situ silylated Au/TS-1-B catalysts
将原位硅烷化改性Au/TS-1-B 催化剂与文献报道的硅烷化改性催化剂进行性能对比,数据见表2。可以明显看出,原位硅烷化改性Au/TS-1-B 催化剂显示出最高的PO 生成速率与氢效,并且该催化剂在200 ℃下反应时仍具有较高的PO 选择性,显示出较好的工业应用前景。
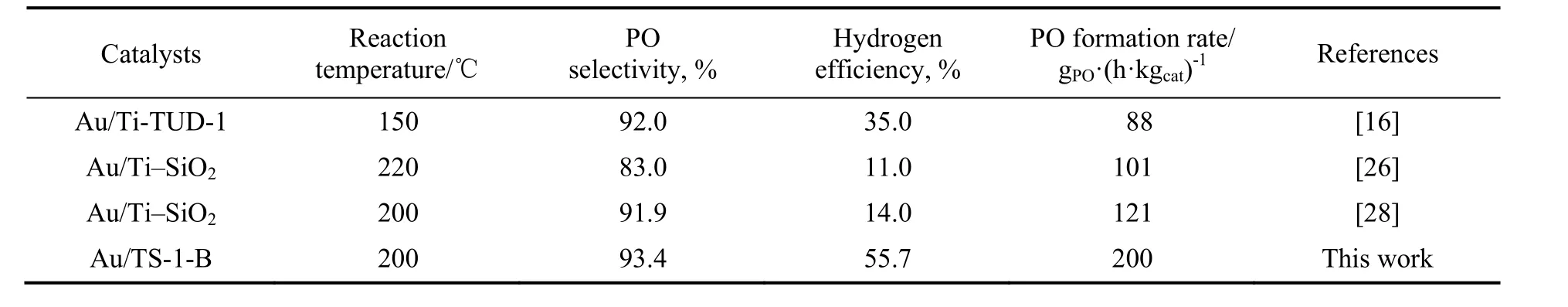
表2 硅烷化改性催化剂的HOPO 反应性能Table 2 Catalytic properties of silylated Au-Ti bifunctional catalysts for HOPO reaction
3 结 论
研究了在Au/TS-1-B 催化剂高温还原过程中通入HMDSO 进行原位硅烷化改性对催化剂结构与性能的调控机制。相比于未改性Au/TS-1-B 催化剂,原位硅烷化改性Au/TS-1-B 催化剂上纳米金颗粒的平均粒径更小。并且原位硅烷化改性提高了Au0的电子结合能,使得原位硅烷化改性催化剂上氢气的转化率明显提高。此外,原位硅烷化改性还显著降低了Au/TS-1-B 催化剂表面的Si—OH 数量,有助于加快反应中生成的H2O 及PO 在催化剂表面的脱附,从而提高了单位时间内催化剂表面暴露Ti—OH位点的数量。因此,原位硅烷化改性对Au/TS-1-B 催化剂上Au—Ti—Si 位点的协同改性显著提高了催化剂的活性。
猜你喜欢
幼儿100(2024年19期)2024-05-29 07:43:34
扬子江诗刊(2023年3期)2023-05-06 10:40:14
大众文艺(2022年16期)2022-09-07 03:08:04
陶瓷学报(2020年5期)2020-11-09 09:22:48
纺织科学与工程学报(2020年1期)2020-06-12 09:14:42
农药科学与管理(2019年5期)2019-08-13 00:48:02
天然产物研究与开发(2018年5期)2018-06-13 03:23:54
当代化工研究(2016年7期)2016-03-20 16:21:55
上海塑料(2015年3期)2015-02-28 14:52:05
河南科技(2014年12期)2014-02-27 14:10:29
