
紫果西番莲果皮花色苷鉴定及其生物活性
2020-07-13 11:48孔钰婷宋洪波安风平
食品科学 2020年11期
何 丹,孔钰婷,宋洪波,2,*,安风平,黄 群,2
(1.福建农林大学食品科学学院,福建 福州 350002;2.福建农林大学 福建省特种淀粉品质科学与加工技术重点实验室,福建 福州 350002)
西番莲(Passiflora edulis)原产于南美洲,在许多热带和亚热带国家均有种植,是最受欢迎的浓郁芳香水果之一,近年来在我国广西、广东、江西及福建等省已规模种植。西番莲具有丰富的营养成分及促健康作用[1],目前其加工以果汁和浓缩果汁为主[2],果皮(占果实质量的50%~55%)通常被废弃。研究报道,紫果西番莲果皮中含有丰富的果胶、膳食纤维、黄酮类化合物及花色苷,其花色苷含量比蓝莓、桑葚和葡萄等水果高[3-5]。花色苷是天然色素的主要成分之一,可赋予水果、蔬菜和花卉等植物蓝色、红色和紫色[6]。研究表明,花色苷能够改善胰岛素抵抗和保护β细胞[7],作用机制主要与其抗氧化活性有关,也可能与对酶的抑制作用或其他途径存在关联[8];此外,花色苷还能够抑制和减少促炎细胞因子的分泌,使其具有一定的抗炎[9]和抗糖尿病活性[10]。本研究旨在明确紫果西番莲果皮中花色苷的组成,评估其中花色苷的体外抗氧化活性及其对2型糖尿病相关酶活力的抑制作用,通过模拟胃肠道消化对其生物利用度进行初步研究,以期为紫果西番莲果皮中花色苷的开发利用提供科学依据。
1 材料与方法
1.1 材料与试剂
‘紫香1号’紫果西番莲购于福建省龙岩市漳平市拱桥镇。
无水乙醇、盐酸、氯化钾和无水乙酸钠(均为分析纯)国药集团化学试剂有限公司;矢车菊素-3-葡萄糖苷(cyanidin-3-glucoside,C3G) 法国Extrasynthese公司;D101型大孔吸附树脂 艾美科健(中国)生物医药有限公司;α-淀粉酶(50 U/mg)、α-葡萄糖苷酶(50 U/mg)、胃蛋白酶(30 000 U/mg)、胆汁盐、胰蛋白酶(2 500 U/mg)、1,1-二苯基-2-三硝基苯肼(1,1-diphenyl-2-picrylhydrazyl,DPPH)、对硝基苯基-α-D-吡喃葡萄糖苷(p-nitrophenylα-D-glucopyranoside,PNPG)、3,5-二硝基水杨酸(3,5-dinitrosalicylic acid,DNS) 上海源叶生物有限公司;2,2’-联氮-双(3-乙基苯并噻唑啉-6-磺酸)二铵盐(2,2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)diammonium salt,ABTS) 阿拉丁试剂(上海)有限公司。
1.2 仪器与设备
TGL-16型台式高速冷冻离心机 四川蜀科仪器有限公司;1260型高效液相色谱仪、6520型高效液相色谱-四极杆飞行时间-串联质谱(high performance liquid chromatography quadrupole-time-of-flight-tandem mass spectrometry,HPLC-QTOF-MS/MS) 美国安捷伦公司;UV-1780紫外-可见分光光度计、Inertsustain C18柱(250 mm×4.6 mm,5 μm) 日本岛津公司;定制玻璃层析柱(170 cm×5 cm) 福州昱安正茂玻璃仪器有限公司。
1.3 方法
1.3.1 紫果西番莲果皮的处理
将紫果西番莲果皮用40 ℃热风干燥至水分质量分数为(8.0±0.5)%,粉碎后过40 目(0.45 mm)筛,于-18 ℃避光保存备用。
1.3.2 紫果西番莲果皮花色苷的提取
称取100 g样品与5 L体积分数60%乙醇溶液(含体积分数0.1% HCl)于烧杯中混合,保鲜膜封口,于黑暗环境下提取24 h,(0.097±0.005)MPa减压抽滤进行固液分离,固体残渣在上述条件下重复提取3 次,合并滤液。滤液于2 555×g、20 ℃离心10 min,(38±1)℃减压浓缩((0.097±0.005)MPa)并回收乙醇,将浓缩液冷冻干燥,即为粗提物。于-18 ℃避光保存备用。
1.3.3 紫果西番莲果皮花色苷的纯化
用蒸馏水将花色苷粗提物配制质量浓度为(17.35±0.62)mg/L的溶液。层析柱填料为D101型大孔吸附树脂,床体积为2 L;上样流速5 mL/min,泄漏量为10%时停止上样;先用10 L、pH(2.5±0.05)的HCl溶液以10 mL/min的流速进行洗脱;再用4 L、pH(2.50±0.05)体积分数90%乙醇溶液以5 mL/min的流速进行洗脱,收集洗脱液。将洗脱液于(38±1)℃下减压浓缩((0.097±0.005)MPa),冷冻干燥制得纯化样品。
1.3.4 紫果西番莲果皮总花色苷含量测定
采用美国分析化学家协会分析方法中的pH示差法测定紫果西番莲果皮花色苷粗提物和纯化样品中的总花色苷含量。
1.3.5 HPLC-二极管阵列检测紫果西番莲果皮花色苷中C3G的含量
用甲醇配制质量溶度为1 mg/mL的样品溶液,过0.45 μm滤膜待测。色谱条件:色谱柱:C18柱(250 mm×4.6 mm,5 μm);流动相A:体积分数0.3%甲酸-水溶液,流动相B:乙腈;洗脱梯度:0~10 min、13%~25% B;10~12 min、25%~20% B;12~22 min、20%~13% B;检测波长520 nm;柱温25 ℃;流速1 mL/min;进样量10 μL;外标法定量。
1.3.6 HPLC-QTOF-MS/MS鉴定紫果西番莲果皮花色苷组分
色谱条件与1.3.5节一致。MS条件:正离子模式;雾化器压力40 psi;干燥器温度350 ℃;气体流量10 L/min;毛细管电压3 500 V;裂解电压135 V;全扫描;扫描范围m/z 50~1 200[11]。
1.3.7 紫果西番莲果皮花色苷体外抗氧化活性测定
1.3.7.1 DPPH自由基清除活性
参照Brand-Williams等[12]的方法,取0.1 mL不同质量浓度的样品溶液(花色苷粗提物、纯化样品和VC)和3.9 mL 0.063 4 mmol/L DPPH-无水乙醇溶液混合,避光反应30 min,用紫外-可见分光光度计于515 nm波长处测定溶液吸光度。空白组以3.9 mL无水乙醇溶液代替DPPH溶液。对照组以0.1 mL无水乙醇代替样品溶液。紫果西番莲果皮花色苷粗提物和纯化样品DPPH自由基清除率按式(1)计算,并计算DPPH自由基半抑制浓度(half inhibitory concentration,IC50)。
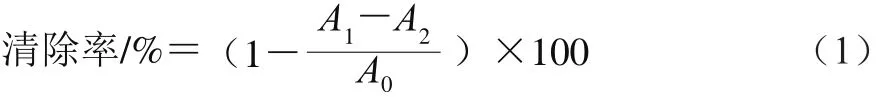
式中:A0为对照组溶液吸光度;A1为样品组溶液吸光度;A2为空白组溶液吸光度。
1.3.7.2 ABTS阳离子自由基清除活性
参照Re等[13]的方法,将20mL 7 mmol/L的ABTS溶液与350 μL 140 mmol/L的过硫酸钾溶液混合,室温下避光反应14 h,制成ABTS储备溶液。取15 μL用甲醇配制的不同质量浓度的样品溶液(花色苷粗提物、纯化样品和VC)与185 μL ABTS储备溶液混合,避光反应30 min,用紫外-可见分光光度计于734 nm波长处测定溶液吸光度。空白组以185 μL甲醇溶液代替ABTS储备溶液,对照组以15 μL甲醇溶液代替样品溶液。根据式(1)计算ABTS阳离子自由基清除率,并计算ABTS阳离子自由基IC50。
1.3.7.3 铁离子还原能力
参照Sarkar等[14]的方法并稍作修改,采用铁离子还原能力(ferric ion reducing antioxidant power,FRAP)法测定。将300 mmol/L pH 3.6的醋酸盐缓冲溶液、10 mmol/L二硫苏糖醇溶液和20 mmol/L FeCl3溶液按体积比10∶1∶1混合配制成FRAP溶液。以FeSO4浓度为横坐标,吸光度为纵坐标,绘制标准曲线,回归方程为Y=0.387 6X+0.184 3(R2=0.999 6),线性范围为0.1~1.5 mmol/L。取0.1 mL不同质量浓度的样品溶液(花色苷粗提物、纯化样品和VC)和3.0 mL FRAP溶液混匀,避光反应4 min后于593 nm波长处测定溶液吸光度。将吸光度代入FeSO4标准曲线,将所得相应的FeSO4浓度定义为FRAP,单位为mmol/L。
1.3.8 紫果西番莲果皮花色苷对α-淀粉酶及α-葡萄糖苷酶的抑制作用
1.3.8.1α-淀粉酶活性抑制率
参考文献[15]中的方法并略作改动。500 μL 1 U/mL的α-淀粉酶溶液与500 μL 1 mg/mL的样品溶液(花色苷粗提物和纯化样品)混合,37 ℃预热10 min,加入500 μL 1 g/100 mL可溶性淀粉溶液(预先溶于pH 6.9的磷酸盐缓冲液(phosphate buffered saline,PBS)并煮沸15 min),37 ℃孵育3 min,加入1 mL DNS显色剂,于100 ℃水浴5 min,冷却后将混合物用超纯水稀释4 倍,于520 nm波长处测定吸光度。以pH 7 PBS代替酶溶液为样品对照,以pH 7 PBS代替样品溶液为空白组;以不添加样品溶液和酶溶液作为空白对照组。按式(2)计算α-淀粉酶活性抑制率。以阿卡波糖为阳性对照,将计算结果除以阿卡波糖对α-淀粉酶抑制率(测定条件相同),紫果西番莲果皮花色苷对α-淀粉酶的抑制率以相对于阿卡波糖的抑制率表示。
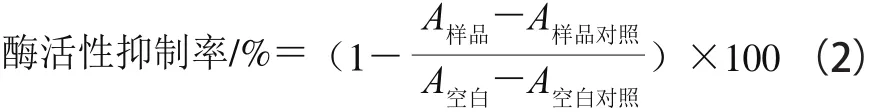
1.3.8.2α-葡萄糖苷酶活性抑制率
参考Mojica等[16]的方法并稍作修改。在96 孔板中加入50 μL 1 mg/mL样品溶液和100 μL 1 U/mL的α-葡萄糖苷酶溶液,37 ℃孵育10 min;加入50 μL 5 mmol/L的PNPG溶液,37 ℃下孵育5 min,于405 nm波长处测定吸光度。按式(2)计算抑制率。以阿卡波糖为阳性对照,将计算结果除以阿卡波糖对α-葡萄糖苷酶抑制率(测定条件相同),紫果西番莲果皮花色苷对α-葡萄糖苷酶的抑制率以相对于阿卡波糖的抑制率(%)表示。
1.3.9 紫果西番莲果皮花色苷的体外生物利用度测定
参考Sengul等[17]的方法并略作改动。将20 mg样品溶解在20 mL pH 1.7的HCl溶液中,加入25.2 mg胃蛋白酶,37 ℃、100 r/min搅拌2 h,再加入500 mg胆汁盐和18 mg胰蛋白酶进行体外模拟消化。向长5 cm的纤维素透析管(截留分子质量12 kDa)中加入5.6 mL 0.75 mol/L NaHCO3溶液,并将其放入消化液中保持2 h。进入管内的溶液代表在体内吸收并进入血清的样品,通过pH示差法分析进入血清样品的总花色苷含量。根据式(3)计算花色苷的体外生物利用度。
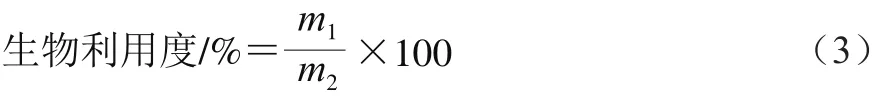
式中:m1为进入透析管内溶液中的花色苷质量/mg;m2为样品中花色苷的初始质量/mg。
1.4 数据处理与分析
每组实验重复3 次,结果用平均值±标准差表示;采用Graphpad prism 8.0.2软件进行单因素方差分析和t检验,P<0.05认为差异有统计学意义。
2 结果与分析
2.1 紫果西番莲果皮中花色苷含量及鉴定
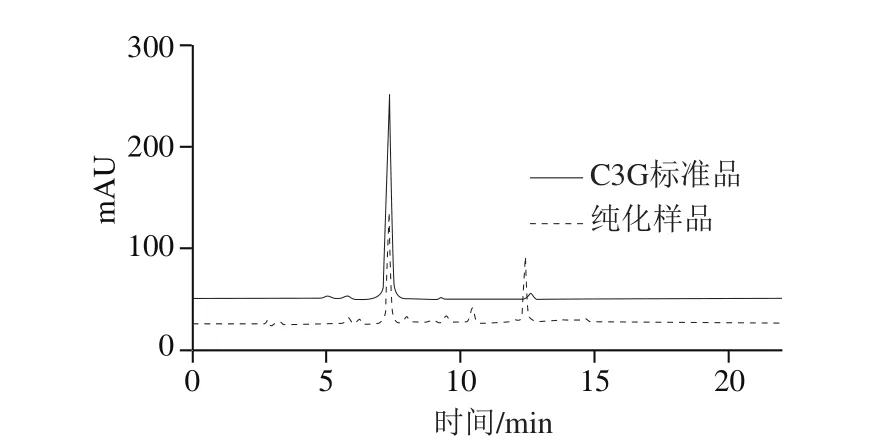
图1 紫果西番莲果皮花色苷纯化样品及C3G标准品的HPLC图Fig. 1 High performance liquid chromatography of purified anthocyanins from Passi flora edulisSims peel and C3G standard
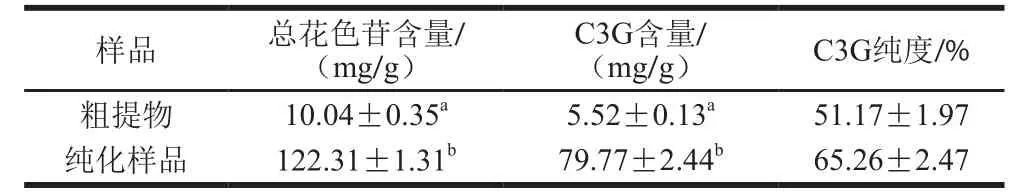
表1 紫果西番莲果皮花色苷的含量及纯度Table 1 Anthocyanin contents and purity of crude and purified anthocyanins from Passi flora edulis Sims peel
紫果西番莲果皮中花色苷粗提物的得率为(3.94±0.11)mg/g。通过比较纯化样品与C3G标准品的保留时间(图1),可以初步推断紫果西番莲果皮中的主要花色苷为C3G,这与文献[17]报道的结果一致。由表1可知,纯化样品中总花色苷含量较粗提物提高了(102.27±1.09)mg/g,回收率为(77.87±0.80)%,C3G纯度由(51.17±1.97)%提高到(65.26±2.47)%。由此表明通过D101型大孔树脂柱层析进行紫果西番莲果皮花色苷的初步纯化是可行的。
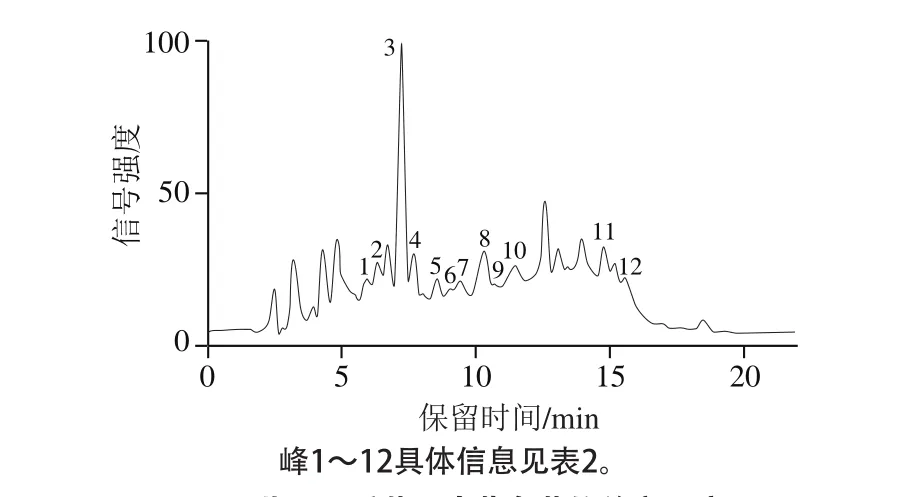
图2 紫果西番莲果皮花色苷的总离子流图Fig. 2 Total ion current chromatogram of anthocyanins in Passiflora edulis Sims peel
采用HPLC-QTOF-MS/MS对纯化样品中的花色苷组分进行结构鉴定。误差质量(δ)作为异核多键相关谱的一个重要特征,按照δ<3对化合物结构进行确认[20],紫果西番莲果皮中花色苷各组分总离子流图如图2所示。理论上,花色苷的糖苷化可以在任意位的羟基上发生,但是某些羟基位置是被优先选择的,如3-OH和5-OH常被糖基取代,而这两者相比时,3-OH会优先被糖苷化[21]。本实验从纯化样品中鉴定出12 种花色苷见表2,结构见图3。
图2中峰1~4和峰8均有m/z 287的糖苷配基离子,表明它们是矢车菊素的糖苷衍生物。根据峰1的分子离子峰M+m/z 773.214 03和碎片离子峰m/z 611.161 27/449.104 09/287.054 29,峰1被鉴定为矢车菊素-3-二葡萄糖苷-5-葡萄糖苷。根据峰2的分子离子峰M+m/z 611.161 51和碎片离子峰m/z 449.112 51(损失m/z 162,葡萄糖)/287.055 91(损失m/z 324,2 分子葡萄糖),因此峰2被鉴定为矢车菊素-3,5-二葡萄糖苷,该物质已在西番莲花冠中被报道[22],但在果皮中鲜见报道。峰3(分子离子峰M+m/z 449.107 56、碎片离子峰m/z 287.054 20)被鉴定为矢车菊素-3-葡萄糖苷。峰4的分子离子峰M+m/z 595.166 40及其碎片离子峰m/z 287.059 20,其损失m/z 308对应于矢车菊素-3-芸香糖苷中芸香糖分子(鼠李糖+葡萄糖),因此峰4鉴定为矢车菊素-3-芸香糖苷。峰3、4所代表物质在西番莲果皮中已见报道[23]。峰8(分子离子峰M+m/z 535.107 50,碎片离子峰m/z 449.106 55/287.054 60)被鉴定为矢车菊素-3-(6”-丙二酰)葡萄糖苷,其在西番莲果皮[18,24]及花冠[22]中均有报道。
碎片离子峰m/z 271表明该物质的糖苷配基为天竺葵素(峰5和峰10)。根据峰5的分子离子峰M+m/z 433.112 50及碎片离子峰m/z 271.06 090(损失m/z 162,葡萄糖),被鉴定为天竺葵素-3-葡萄糖苷,其在紫果西番莲果皮中已有报道[18]。峰10具有2 个特征碎片离子峰,分别为m/z 433.112 60([M]+-146)和m/z 271.060 81([M]+-308),表明糖苷配基与芸香糖(鼠李糖+葡萄糖)连接。因此,峰10被鉴定为天竺葵素-3-芸香糖苷。
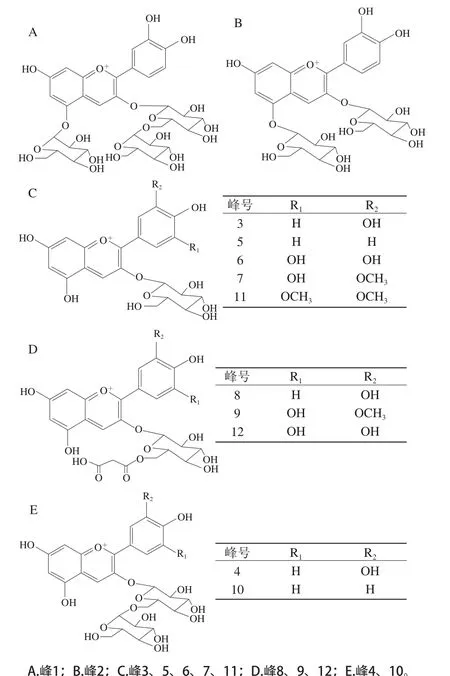
图3 紫果西番莲果皮中花色苷的化学结构Fig. 3 Structures of anthocyanins in Passiflora edulis Sims peel
根据峰6和峰12的分子离子峰及碎片离子峰的特征信息,峰6和峰12分别被鉴定为飞燕草素-3-葡萄糖苷和飞燕草素-3-(6”-丙二酰)葡萄糖苷。峰7及峰9的碎片离子峰表明这两种物质均为芍药素苷(m/z 301)的衍生物。其中峰7的分子离子峰M+m/z 463.124 18,表明其由芍药素与一分子葡萄糖结合而成,所以峰7被鉴定为芍药素-3-葡萄糖苷,其在紫果西番莲果皮中已见报道[18]。峰9(分子离子峰M+m/z 549.124 21,碎片离子峰m/z 463.124 00/301.068 40)被鉴定为芍药素-3-(6”-丙二酰)葡萄糖苷。根据峰11的质谱特征信息,其被鉴定为锦葵色素-3-葡萄糖苷,在西番莲果皮中已见报道[19]。

表2 紫果西番莲果皮花色苷的鉴定及质谱特征Table 2 Identification and mass spectrometric properties of anthocyanins from Passi flora edulis Sims peel
2.2 紫果西番莲果皮花色苷的体外抗氧化活性
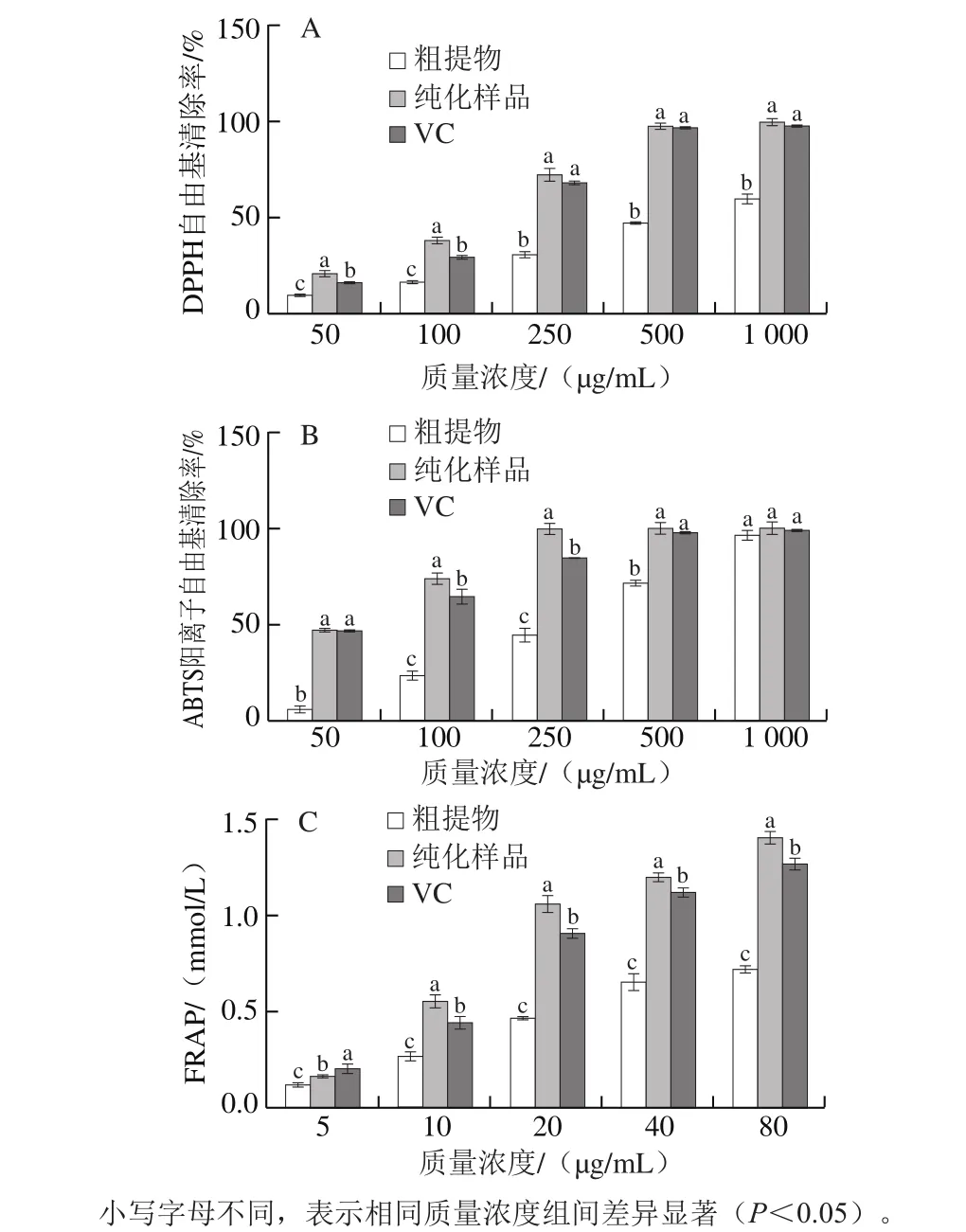
图4 紫果西番莲果皮花色苷和VC的DPP自由基(A)、ABTS阳离子自由基(B)清除活性和FRAP(C)Fig. 4 Scavenging activities of anthocyanins from Passi flora edulis Sims peel and VC on 1,1-diphenyl-2-picrylhydrazyl radicals (A), 2,2’-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) cation radicals (B) and their ferric ion reducing antioxidant power (C)
由图4可以看出,在质量浓度为50~1 000 μg/mL时,紫果西番莲果皮花色苷粗提物、纯化样品和VC的DPPH自由基清除率均随质量浓度的增加而增加;质量浓度为1 000 μg/mL时,花色苷纯化样品的DPPH自由基清除率略高于VC、显著高于粗提物(P<0.05);花色苷纯化样品、VC和粗提物对DPPH自由基的IC50分别为126.6、152.2 μg/mL和604.1 μg/mL。ABTS阳离子自由基清除率的变化趋势与之类似,花色苷纯化样品、VC和粗提物的IC50分别为54.47、58.61 μg/mL和260.2 μg/mL。3 种花色苷样品的FRAP在质量浓度5~80 μg/mL范围内呈增加趋势;在80 μg/mL时,花色苷纯化样品FRAP显著高于VC和粗提物(P<0.05),此时FRAP分别为(1.413±0.051)、(1.277±0.025)mmol/mL和(0.724±0.021)mmol/mL。较高的抗氧化能力通常与花色苷及总酚的含量相关[31-32]。研究表明,覆盆子、蓝莓中的类黄酮化合物(如花色苷)具有优于VC的抗氧化能力[33],可能是由于花色苷的高抗氧化能力与其结构中含有大量酚羟基基团有关[21]。总体而言,本研究发现紫果西番莲果皮花色苷纯化样品的抗氧化能力较粗提物显著提高,且优于VC。
2.3 紫果西番莲果皮花色苷对α-淀粉酶及α-葡萄糖苷酶的抑制作用

表3 紫果西番莲果皮花色苷对α-淀粉酶和α-葡萄糖苷酶的抑制率Table 3 Inhibition rates of crude extract and purified anthocyanins from Passi flora edulis Sims peel against α-amylase and α-glucosidase
抑制与2型糖尿病相关的关键酶(α-淀粉酶和α-葡萄糖苷酶)活力能够减少脂肪吸收和防止餐后血糖升高,是治疗2型糖尿病的早期途径[34]。花色苷通过与这些酶结合,从而影响其活性。Peng Jinming等[35]发现,黑花生皮的花色苷提取物对α-淀粉酶和α-葡萄糖苷酶的抑制作用具有浓度依赖性,不同结构的花色苷与酶的结合能力存在差别。如表3所示,紫果西番莲果皮花色苷粗提物和纯化样品对α-淀粉酶活力抑制率分别为(26.76±0.98)%和(34.11±1.47)%,对α-葡萄糖苷酶活力抑制率分别为(52.87±2.31)%和(60.36±2.05)%,表现出更强的抑制作用。花色苷纯化样品对两种酶活力的抑制作用均显著高于粗提物(P<0.05)。
紫果西番莲果皮花色苷粗提物及纯化样品对α-淀粉酶和α-葡萄糖苷酶的抑制作用均不及阿卡波糖,然而,一些用于治疗肥胖和2型糖尿病的药物(奥利司他和阿卡波糖)易引发如胃肠胀气、腹泻和腹胀等不良反应,因此常规药物与具有生物功效的天然产物联用是减轻药物副作用的新途径[34]。另一方面,糖尿病人由于血糖升高,糖基化终末期产物和自由基明显增多,这些自由基会促进炎症反应发生,这也是糖尿病慢性病变的一个重要原因,因此抗氧化对于预防和延缓糖尿病的慢性并发症具有重要作用[36]。
紫果西番莲果皮花色苷纯化样品具有很好的抗氧化活性和抑制α-淀粉酶及α-葡萄糖苷酶的双重功效,因此在开发降血糖功能食品或药物方面具有极大潜力。
2.4 紫果西番莲果皮花色苷的体外生物利用度

表4 紫果西番莲果皮花色苷粗提物和纯化样品的生物利用度Table 4 Bioavailability of crude extract and purified anthocyanins from Passi flora edulis Sims peel
生物利用度是指有效成分被吸收进入人体循环的比例,描述了口服物由胃肠道吸收以及经过肝脏到达体循环血液中的量占口服剂量的百分比。本实验采用体外模拟评价紫果西番莲果皮花色苷的生物利用度,从表4可以看出,花色苷纯化样品的生物利用度为(9.78±0.31)%,显著大于粗提物的(5.45±0.39)%(P<0.05)。研究表明,覆盆子花色苷粗提物在肠消化过程中的生物利用度为5.3%[37],与本研究粗提物体外模拟消化的生物利用度接近。由此可见,紫果西番莲果皮花色苷纯化样品具有更高的生物利用度。
3 结 论
基于保留时间和二级质谱数据,初步鉴定出紫果西番莲果皮中存在12 种花色苷。花色苷纯化样品的抗氧化活性显著高于粗提物(P<0.05),且优于VC,纯化样品对α-淀粉酶和α-葡萄糖苷酶抑制作用也显著高于粗提物(P<0.05),说明花色苷纯化样品具有很好的抗氧化活性和抑制血糖升高的潜能。花色苷粗提物和纯化样品的体外生物利用度分别为5.45%和9.78%,纯化样品具有更高的生物利用度。紫果西番莲果皮花色苷纯化样品在降血糖、抗氧化功能食品和医药方面具有很好的开发潜力。还需要运用核磁共振波谱等手段进一步解析紫果西番莲果皮花色苷的组成和结构,进而阐明结构与其强抗氧化性之间的关系,明确花色苷与纯化样品中其他成分可能存在的协同增效作用。
猜你喜欢
中草药(2022年20期)2022-11-15
现代食品科技(2022年9期)2022-10-09
中国食品学报(2022年8期)2022-09-07
江苏农业学报(2022年4期)2022-09-07
热带作物学报(2022年5期)2022-06-01
中国果业信息(2021年5期)2021-12-05
化工设计通讯(2021年2期)2021-01-07
农民致富之友(2020年6期)2020-04-08
农产品加工(2019年3期)2019-01-06
中学生物学(2018年6期)2018-01-17
