
镍铜合金立方体纳米晶的脉冲电沉积及电催化析氢性能
2020-07-06 01:52:02孙强强曹宝月周春生张国春王增林
高等学校化学学报 2020年6期
孙强强, 曹宝月, 周春生, 张国春, 王增林
(1. 商洛学院化学工程与现代材料学院, 陕西省尾矿资源综合利用重点实验室, 商洛 726000;2. 陕西师范大学化学化工学院, 应用表面与胶体化学教育部重点实验室, 西安 710119)
氢能是一种环保、 可再生的二次能源. 电解水制氢[1~3]因为原料来源广、 碳排放低、 环境友好且可循环利用, 被认为是通向氢经济的最佳途径. 但电解水过程阴阳两极析氢(HER)和析氧(OER)过电位的存在造成了极大的电能消耗. 为推进电解水制氢的工业化进程, 设计高活性、 环境友好的电催化剂是电解水制氢产业发展的关键. 工业上普遍采用商业Pt/C等贵金属电极[4]作为电解水的阴极, 而贵金属的稀缺性及高成本限制了其在电解水制氢工业中的规模化应用. 近年来, 价格低廉且储量丰富的第一过渡系金属及其合金、 磷化物, 硫化物、 硒化物、 氮化物、 碳化物等[5~7]被广泛应用到析氢反应中. 多元过渡金属基催化剂因其理想的双金属协同催化作用、 优异的电子电导率及抗腐蚀性、 稳定的化学结构以及低廉的成本而成为电催化高效析氢的热门材料. 由于具有优异的析氢本征活性, 镍基合金电极作为析氢电极广受科研工作者的青睐, 如MoNi4, Ni-W, Ni-Fe及Ni5P4等[8~10]. 根据Brewer-Engel价键理论[11], 铜的d电子与镍的半空d轨道结合形成金属间的协同电催化作用, 有助于提高析氢反应的本征催化活性; 而且比与商业Pt/C更低廉的成本及丰富的储量使多元镍铜合金电极[12,13]成为替代商业Pt/C应用于工业化制氢最有前景的电极材料.
局限于暴露出的有限的催化活性位点, 现有镍铜电极材料的析氢活性与商业Pt/C相比仍有较大差距, 对镍铜金属间的协同效应也缺乏深入的理解[14~17]. 选择三维多孔结构的泡沫镍(NF)为基板, 通过引入由互联微孔交织而成的三维立体空间可增加结构复杂度, 促进物料传递及气体扩散, 从而减小在高电流密度下的能量损耗. 表面成孔技术[18]可在泡沫镍表面构筑纳米多孔薄膜, 增大电催化活性面积, 暴露大量电催化活性位点, 改善析氢性能. 为增加单位催化位点的固有活性, 元素掺杂或构筑异质界面也是改善析氢活性的有效办法.
脉冲电沉积是一种以高频率的断续电流来制取金属合金薄膜的沉积技术[19,20], 相比传统直流电沉积, 因其脉冲宽度窄, 峰电流密度大, 可以大幅降低电沉积过程的浓差极化. 利用脉冲电沉积制备纳米晶体可以提高材料的延展性、 耐磨耐腐蚀性, 避免金属氧化物的形成, 增强镀层的附着力, 获得更加细致均匀的镀层, 增强材料的催化活性及稳定性. 本文以泡沫镍为基板, 采用常规脉冲电沉积法在泡沫镍表面构筑二元镍铜合金体相, 制备了具有多级复合纳米尺寸的镍铜基电催化剂, 并研究了其电催化析氢性能.
1 实验部分
1.1 试剂与仪器
硫酸镍、 硫酸铜、 硼酸、 氢氧化钾、 氢氧化钠、 盐酸和无水乙醇均为分析纯, 购于国药集团化学试剂有限公司; 超纯水为自制.
CHI 760E电化学工作站, 上海辰华仪器有限公司; SU-8020冷场发射扫描电子显微镜(FESEM, 二次电子成像, 加速电压10 kV), 日立高新技术公司; Tecnai G2 F20冷场发射透射电子显微镜(TEM, 工作电压200 kV), 美国FEI公司; Bruker D8 Discover高分辨X射线衍射仪(XRD, 电压40 kV, 管电流40 mA, CuKα辐射源,λ=0.15418 nm, 2θ=30°~80°, 扫描速率为8°/min), 德国布鲁克科技有限公司; PHI-5000 X射线光电子能谱仪(XPS, 工作电压15 kV, AlKa射线), 美国ULVCA-PHI公司; in Via Reflex显微共聚焦拉曼光谱仪(Raman, 测试波长532 nm), 英国雷尼绍公司.
1.2 镍铜合金电极的制备
将纯度为99.9%、 厚度为1.5 mm的金属泡沫镍裁剪成2.0 cm×0.5 cm的泡沫镍条, 分别在5 mol/L的盐酸和95%(体积分数)的乙醇溶液中超声浸泡15 min, 除去表面的氧化物及表层油污, 再用去离子水清洗3次后储存于乙醇溶液中, 每次使用前真空干燥. 以预处理后的泡沫镍为工作电极, 平板镍片为对电极, 饱和甘汞电极(SCE)为参比电极, 采用常规脉冲伏安法在标准三电极体系中进行一步电沉积(电解液组成为0.05 mol/L NiSO4+0.0075 mol/L CuSO4+0.05 mol/L H3BO3, 用NaOH溶液调节溶液的pH值至4.0; 沉积参数: 起始电位-1.0 V, 终点电位-1.5 V, 电位增量0.004 V, 脉冲宽度0.2 s, 工作周期 2.0 s, 占空比均为10%)即可制得NiCu/NF电催化剂, 于空气中自然干燥后备用.
1.3 电化学测试
在室温下, 采用标准的三电极体系在CHI760E电化学工作站上进行镍铜合金电极的电化学性能测试, 以制备电极为工作电极(有效工作面积0.70 cm2), 饱和甘汞电极(SCE)为参比电极, 石墨棒为对电极, 电解质溶液为1 mol/L KOH. 采用线性扫描伏安法(LSV)测定电催化剂的析氢性能, 析氢过程扫描电位窗口为-0.8~-1.5 V, 扫描速度为2 mV/s, 极化曲线均经过95%电阻补偿. 多电流阶跃测试分10个阶段进行, 自50 mA/cm2逐段增至500 mA/cm2, 每500 s电流密度递增50 mA/cm2. 采用计时电位法分别测试电流密度为10和100 mA/cm2时电极析氢电位随时间的变化情况, 测试时间为24 h, 根据析氢过程电位的波动幅度来评价制备电极的长时稳定性. 在非法拉第感应窗口-0.05~0.05 V下, 测试NiCu/NF电极在不同扫速(5, 10, 25, 50, 100, 200 mV/s)下的充电双电层库仑曲线, 依据充电电流(ic)与扫描速率(v)的直线关系, 获得制备电极的双电层电容Cdl(mF/cm3), 按下式换算后即可求得相应的电化学活性面积(ECSA):
ECSA=Cdl/Cs(1)

采用电化学交流阻抗谱(EIS)分析电极表面的动力学控制过程. 测试参数为: 测试电位为-1.250 V(vs. SCE), 扫描频率为0.1~105Hz, 振幅为5 mV. 测试完毕, 通过ZSimp Win软件选择合适的等效电路图进行拟合, 即可获得溶液电阻(Rs)、 电荷转移电阻(Rct)等数据, 从而分析电极表面的动力学控制过程. 在析氢活性的归一化处理[22]中将析氢极化曲线中测得的电流密度j(mA/cm2)按下式换算为校正电流密度jc(mA/cm2)后, 以jc对电位E(V,vs. RHE)绘图, 获得校正后的极化曲线.
jc=j/Δn(2)
式中: Δn为制备电极电化学活性面积相对于基板NF的倍数, 可由下式求得:
Δn=ECSAt/ECSAs(3)
式中: ECSAt(cm2)和ECSAs(cm2)分别为制备电极与基板NF的电化学活性面积.
2 结果与讨论
采用常规脉冲电沉积法制备镍铜合金电催化剂时, 在合适的电位窗口下, 电流导通过程金属镍铜在导电基板表面发生共沉积. 在晶体生长中, 铜比镍优先成核, 且生长速率快, 在基底表面沿特定晶向择优生长后, 便会形成以铜为核, 镍为壳的规则多面体结构[23], 为后期在泡沫镍表面构筑纳米阵列奠定基础. 电沉积过程的导电基板、 沉积参数、 电解液浓度等直接影响金属的电结晶行为[24], 进而影响合金薄膜的形貌及电催化性能, 制备方案及参数的优化过程见图S1~图S4(见本文支持信息). 优选结果如下: 电解液组成为0.05 mol/L NiSO4+0.0075 mol/L CuSO4+0.05 mol/L H3BO3, 用NaOH溶液调节溶液的pH值至4.0, 以NF为基板, 采用常规脉冲伏安法电沉积, 起始电位 -1.0 V, 终点电位-1.5 V, 电位增量0.004 V, 脉冲宽度0.2 s, 工作周期 2.0 s, 占空比均为10%.
2.1 镍铜合金电催化剂的物理表征
通过SEM, EDX, TEM及XRD对优选NiCu/NF的表观形貌、 组成及元素分布进行测定, 结果如图1和图S5(见本文支持信息)所示.

Fig.1 SEM images(A, B) and high angle circular dark field(HAADF) SEM image(C), mapping images ofNi(D), Cu(E), D(F) of the optimal NiCu/NF
由图1(A)可知, NiCu/NF很好地保留了泡沫镍的三维网状多孔结构, 获得的镍铜薄膜均匀地生长在泡沫镍的韧带上, 表明其与泡沫镍有良好的相容性, 其间较强的附着力能保证其在催化过程优异的稳定性. 从图1(B)发现, 镍铜薄膜是由平均粒径约为70 nm的切角立方体结构排列或堆积而成. 分析认为, 这是因为硼酸小分子择优吸附在镍铜的(111)晶面, 使晶体沿<111>方向生长速率加快, 而沿<001>方向的生长速率减慢, 最终形成立方体结构的镍铜合金, 暴露于台阶位和边缘位的高指数面原子能在电催化过程中表现出高的催化活性[25]. 泡沫镍的三维网状微孔结构与镍铜合金的立方体纳米晶构成了NiCu/NF二级分层复合微纳结构, 可为后续电催化反应中的物料传递及气体逸出提供全方位的扩散通道. 从组成分析可知, 泡沫镍表面的镍铜薄膜由Ni, Cu, O 3种元素组成(图S5). 由NiCu/NF的面扫分布图[图1(D~F)]发现, Ni元素分布在纳米晶粒边缘区域, Cu元素分布于立方体纳米晶的体相中心区域, 而O元素则均匀分布在整个表面. O的存在是因为纳米颗粒暴露在空气中发生表面氧化的缘故.
为进一步探究镍铜薄膜的微观结构、 晶型及物相, 将镍铜合金薄膜从泡沫镍表面超声剥离后, 测试了其TEM照片及XRD谱图, 结果见图2.
图2(A)中平均尺寸约为70 nm的规则立方体结构进一步验证了镍铜合金薄膜的表观形貌. 从HRTEM照片[图2(B)]中可以观察到大量密集排列、 清晰的晶格条纹, 其中纳米颗粒内部晶面间距为0.204, 0.178和0.208 nm的晶格条纹分别归属于金属Ni的(111), (200)晶面和金属Cu的(111)晶面, 而颗粒表层晶面间距为0.241 和0.209 nm的晶格条纹分别归属于NiO的(111)和(200)晶面[26]. 在选区电子衍射图[SAED, 图2(C)]中可以观察到由大量离散斑点所构成的圆环, 表明获得镍铜薄膜呈多晶形态. 根据倒易空间标尺的换算, 离散斑点对应晶格间距的d值分别为0.204, 0.178, 0.125 nm及0.209, 0.181, 0.128 nm, 分别归属于金属Ni和Cu的(111), (200)及(220)晶面[27,28]. HRTEM及SAED 分析均表明多晶的镍铜薄膜由金属Ni, Cu及NiO物相构成, 而NiO相的形成则主要是由立方体结构的镍铜纳米晶具有较高的活性, 暴露于空气中表层的活性镍被局部氧化所致. 薄膜的XRD谱图[图2(D)]中分别出现了金属Ni和Cu的(111), (200), (220) 3个晶面的特征衍射峰, 进一步验证了镍铜薄膜主晶相为金属态Ni和Cu, 且3组分离而尖锐的衍射峰表明两者以独立分相形式存在. 谱图中未出现NiO的特征衍射峰, 这是因为合金薄膜表层的活性镍发生局部氧化, 仅形成了一层纳米级别、 含量较低的氧化物薄膜, 故未显著出峰.

Fig.2 TEM(A) and HRTEM(B) images, SAED(C) and XRD(D) patterns of NiCu/NF
为确定NiCu/NF组成元素的价态, 测定了该电极材料的XPS及Raman谱图(图3). Ni2p的高分辨XPS谱[图3(A)]在结合能为852.8和870.2 eV处出现了一对分裂能为18.0 eV的微弱的自旋轨道峰, 分别归属于Ni(0)的2p3/2和2p1/2轨道, 表明电极材料表面存在金属态Ni. 而在结合能为855.4和873.4 eV处出现了一组宽大的Ni2p自旋轨道峰, 同时在861.1和880.2 eV处还出现了相对应的伴峰, 这表明除了金属态的Ni, 材料表面的镍还以Ni(Ⅱ)[29]形式存在, 这与TEM分析结果一致. Cu2p的高分辨XPS谱[图3(B)]在932.8和952.8 eV处出现了Cu2p的自旋轨道峰2p3/2和2p1/2, 两者的分裂能达到20.0 eV, 这与金属Cu的自旋轨道峰位置相符, 表明电极表面的Cu元素以Cu(0)形式存在[30]. 在O1s的高分辨谱图[图3(C)]中有两个光电子发射峰, 表明O有两种键合形式. 结合能为531.5 eV的发射峰归属于吸附水分子中的羟基氧, 结合能为529.6 eV处的发射峰则对应于NiO中的Ni—O键[31], 这还可以从NiCu/NF的Raman光谱图中得以验证. 在图3(D)中, 基板NF未出现明显的拉曼峰, 而NiCu/NF在531 cm-1处出现了一个显著的拉曼峰, 可归属于NiO中Ni—O键的特征伸缩振动峰[32]. TEM, XPS表征以及Raman分析证实了电极材料中活性镍表层NiO晶相的形成.

Fig.3 XPS spectra of Ni2p(A), Cu2p(B), O1s(C) and Raman spectrum(D) of NiCu/NF electrocatalyst
2.2 镍铜基电催化剂在析氢反应中的电化学性能
为评价镍铜电催化剂在析氢反应中的电化学性能, 测定了其在析氢过程的催化活性、 动力学特性及催化稳定性, 结果见图4.

Fig.4 LSV polarization curves(A), Tafel plots(B), multi-current process(C) and chronoamperometric curves(D) of NiCu/NF towards HER (C) The step of current density is 50 mA/cm2.
图4(A)出了NF, Ni/NF及NiCu/NF电极在析氢过程中的LSV极化曲线. 由图可知, 在电流密度为10 mA/cm2时, NiCu/NF的析氢过电位仅为86 mV, 虽比贵金属催化剂Pt/C/NF的30 mV大, 却远小于同条件下Ni/NF的160 mV及基板NF的212 mV. 表明泡沫镍表面镍铜合金立方体纳米微晶的构建大幅促进了材料析氢活性的提升, 使NiCu/NF在HER中表现出优异的催化活性. 这除了取决于镍铜金属间的协同效应, 还与形成的二级微纳结构有关. NiCu/NF的析氢活性显著优于文献报道的多数非贵金属析氢催化剂, 如NiFeOx/CFP(88 mV)[33], o-CoSe2|P(104 mV)[34], S-MoP(104 mV)[35], NiFeV/NF(125 mV)[36]及Ni5P4/Ni(150 mV)[37]等.
为了评价镍铜合金电催化剂在析氢反应中的动力学特性, 测试了其在HER过程的塔菲尔(Tafel)曲线. 由图4(B)可知, 在电催化析氢过程中, NiCu/NF电极的Tafel斜率为56.9 mV/dec, 虽高于Pt/C/NF的34.6 mV/dec, 却显著小于同条件下Ni/NF的91.5 mV/dec及基底NF的123.0 mV/dec, 表明NiCu/NF在HER中同样表现出优异的动力学特性, 这与催化过程电极表面的电子传递能力有关. Tafel斜率是评价析氢反应动力学及反应机理的重要参数, 可用于判定电极表面析氢反应的决速步骤. NiCu/NF电极在析氢过程的Tafel斜率为56.9 mV/dec, 表明Volmer-Heyrovsky反应为NiCu/NF电极表面析氢过程的决速步骤, 即反应速率由金属表面吸附氢原子的电化学脱附过程决定[38,39].
电极材料的稳定性是评价电催化剂工业应用价值的重要依据. 图4(C)是NiCu/NF在碱性溶液中的多电流阶跃曲线, 反映了电极在分段增加的电流密度下实际电位随时间的变化情况. 结果发现, 在每个阶跃的电流密度下, 电位值几乎保持不变, 即使在高电流密度(500 mA/cm2)下响应曲线仍能保持水平, 表明NiCu/NF在析氢过程中表现出优良的导电性、 快速电荷传递、 超强的气体扩散能力和传质能力以及高的机械强度, 进而表明其具有优异的析氢稳定性.
图4(D)为NiCu/NF在电流密度分别为10和100 mA/cm2时的计时电位曲线. 初始时, 由于电极的活化及吸附气泡短暂的集聚, 使得高电流密度(100 mA/cm2)下的析氢电位有所增加. 待电极稳定后, 催化水分解析氢反应在24 h内电势基本保持不变. 在电流密度为10和100 mA/cm2时, 电势始终分别保持在-0.05 和-0.18 V(vs. RHE), 而且在连续24 h的稳定性测试中, 相应的电位波动仅为12 和20 mV, 表明NiCu/NF在长时间的HER过程中优异的耐用性及超高的机械强度, 这为其在电解水制氢领域的工业应用奠定了基础.
2.3 NiCu/NF电催化剂的高催化活性分析
ECSA的大小反映了电极表面暴露出的催化活性位点的多少, 也直接决定了电极材料催化活性的强弱. 电化学交流阻抗谱(EIS)是评价固相电极与液相电解质界面特性的重要手段, 可以反映催化剂表面的动力学规律[40]. 为探究NiCu/NF高催化活性及优异动力学特性的原因, 采用循环伏安法、 电化学交流阻抗法和归一化法分别测定了NiCu/NF在析氢过程中的双电层电容、 电荷转移电阻以及ECSA校正后的LSV极化曲线, 结果见图5.

Fig.5 CV(A) and double-layer capacitance(B) curves of NiCu/NF towards HER, Nyquist plots(C) and LSV polarization curves(D) by ECSA normalization of NF, Ni/NF and NiCu/NF towards HER Inset of (C): equivalent circuit diagrarn.
图5(A)和(B)分别为NiCu/NF电极在析氢过程中的CV曲线及充电双电层库仑曲线. 根据图中电位为0时充电电流(jc)与扫描速率(v)的线性关系, 求得NiCu/NF电极的双电层电容Cdl为15.03 mF/cm2, 根据式(1)换算后相应的ECSA为375.8 cm2. 同时, 比较了相同条件下Ni/NF(8.11 mF/cm2和202.8 cm2)和空白NF(0.97 mF/cm2和24.3 cm2)的Cdl和ECSA. 研究发现, NiCu/NF的ECSA是空白NF的15.5倍, 这是因为NiCu/NF电催化剂的二级分层结构保证了固相界面与电解质溶液充分接触, 促进了两相间的物料传递, 更可为气体产物的逸出提供充足的通道, 立方体结构的纳米阵列使其在HER中暴露出大量的活性位点.
图5(C)为NF, Ni/NF及NiCu/NF电极在析氢过程中的Nyquist曲线. 利用插图中的等效电路图对Nyquist 曲线进行拟合, 发现三者在测试频率区域均呈现规整的半圆弧, 这主要源于在固相电极与电解液两相界面上水分子得电子发生还原过程的电荷转移电阻Rct, 表明电极表面HER动力学过程均受电化学反应控制[41]. 等效电路图中的Rs为导线电阻、 电极夹电阻以及溶液电阻三者的串联电阻, CPE为具有电容性质的常相位元件. 从NF, Ni/NF到NiCu/NF,Rct由68.2, 9.4 Ω/cm2减小至6.3 Ω/cm2, 这要归因于NiCu/NF电极独特的二级分层微纳结构所带来大量的反应通道及活性接触点, 使两相界面上的电荷传输具有较小的阻力. 随着镍铜合金的复合, 电极材料的导电性增强, 电荷传输速率也随之增大. 同时, 立方体纳米晶的构建增强了法拉第过程的界面接触, 促进了电极表面电荷转移及传质过程, 使析氢反应具有较快的动力学速率, 这有利于提高单位电极表面积内的催化能力.
电极材料的催化活性除了取决于材料表面催化活性位点的数量, 还取决于材料表面单位活性面积催化位点的固有活性. 前者可以通过测定电催化剂的ECSA定量评价, 后者则需要通过析氢活性的归一化处理来衡量, 以ECSA相对于空白NF的增加倍数对析氢极化曲线进行校正, 排除催化剂结构特性所引起催化活性位点增加的影响, 实现对催化剂表面活性位点的本征活性的评价. 为此, 比较了Ni/NF和NiCu/NF单位催化活性位点的本征活性, 校正后的极化曲线及相应的过电位如图5(D)所示. 结果发现, 在电流密度为10 mA/cm2时, 归一化处理后Ni/NF电极的析氢过电位为207 mV, 与空白NF的212 mV仅相差5 mV, 表明Ni/NF的催化活性完全来自于结构优势所引起的ECSA的增加. 而NiCu/NF电极的析氢过电位为171 mV, 相比同条件下的NF下降了41 mV, 证实了铜的引入会增强电极的本征析氢活性.
为探究镍铜对HER产生协同催化作用的原因, 比较了NiCu/NF与基准镍粉不同晶面的特征衍射峰位置. 如图6(A)~(C)所示, 镍铜薄膜中金属Ni相的3个特征衍射峰与基准Ni相比均向小角度方向偏移(2θ平均向左偏移0.22°), 这表明镍铜在合金化过程中部分Cu原子进入富Ni体相中[16]. 根据前期研究成果[21], Cu原子进入Ni的晶格中发生取代后将引起晶格畸变, 增强邻近Ni原子对活性氢原子的吸附能力, 优化其吸/脱附平衡, 进而提升镍基材料的本征析氢活性.
为探究立方体镍铜纳米晶表层NiO薄膜对析氢反应的影响, 将制得的NiCu/NF在氩气和氢气气氛中中温还原处理, 测定了H2还原前后制备电极的析氢极化曲线, 结果如图6(D)所示. 从组成分析看, H2还原后NiCu/NF中O的摩尔分数由原来的7.12%降至0.25%, 表明表层氧化物几乎被完全还原. 在电流密度为10 mA/cm2时, 还原后NiCu/NF的析氢过电位为103 mV, 较还原前(86 mV)增大了17 mV, 表明NiCu/NF表层NiO薄膜的形成确实有助于析氢活性的提升. 分析认为, 活性镍表层氧化镍的形成促使其与体相镍之间形成了NiO/Ni异质结, 增强了邻近Ni原子表面产生的H2分子的脱附能力, 进而提升电催化材料的本征析氢活性[42]. 总之, 镍铜电催化剂二级复合立方体纳米阵列的结构特性、 金属镍铜的协同效应以及氧化镍/镍异质界面的形成共同促进了电极材料催化活性的大幅提升, 使NiCu/NF展现出优异的电催化活性及稳定性.
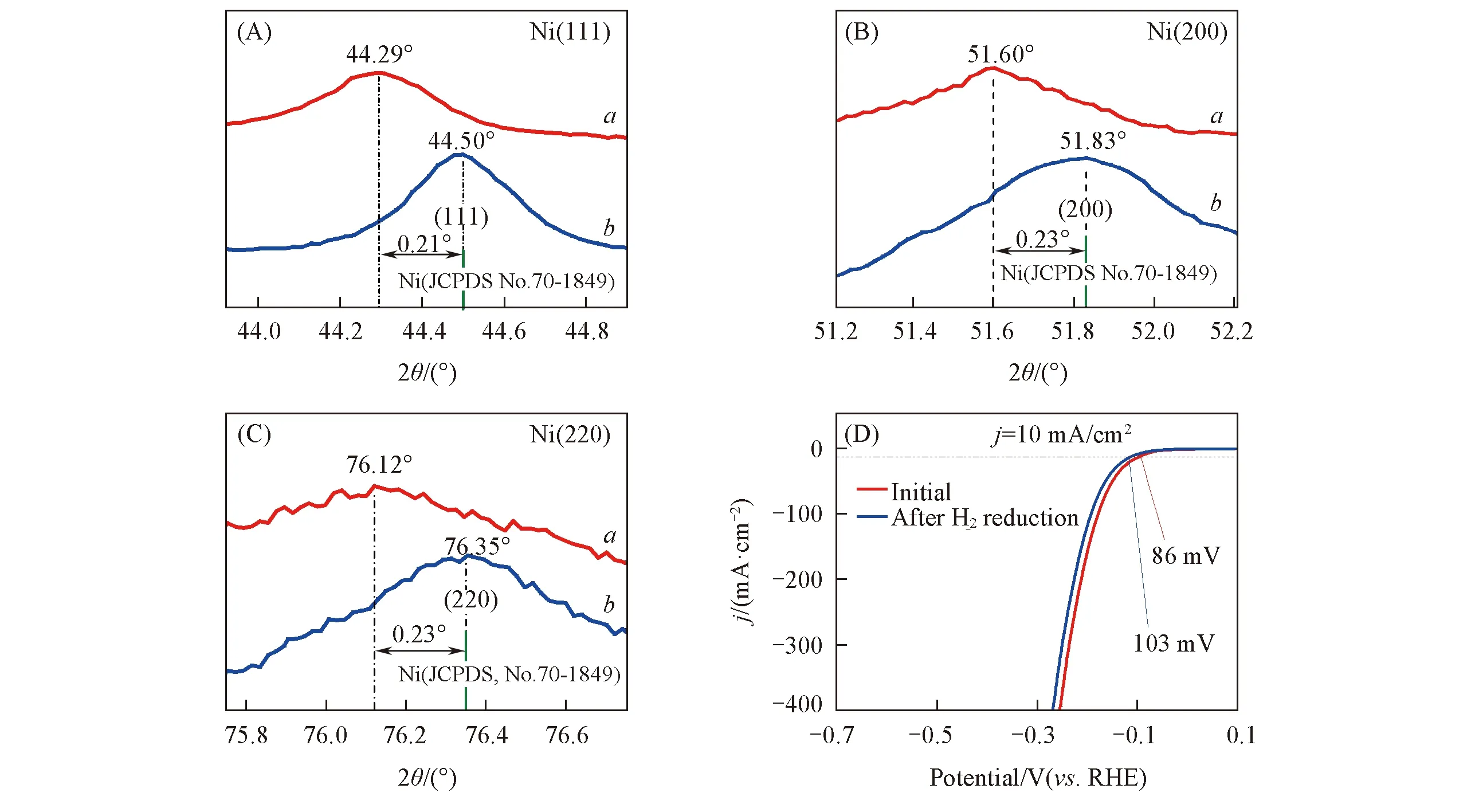
Fig.6 Comparision of characteristic diffraction peaks of (111)(A), (200)(B), (220)(C) planes of Ni phases in NiCu films(a) and standard Ni powder(b) and LSV polarization curves of NiCu/NF before and after H2 reduction(D)
3 结 论
采用常规脉冲伏安法在泡沫镍表面构建了立方体结构的镍铜合金电催化剂. 研究发现, 主晶相为金属态Ni, Cu的催化剂中, Ni, Cu元素以独立分相形式存在; 平均粒径为70 nm的立方体镍铜纳米晶暴露在空气中, 表层形成了纳米级别的NiO薄膜. 二级复合微纳结构的形成使NiCu/NF的ECSA约为NF的15.5倍. 铜的引入一方面通过增大电极材料的导电性增大了电极表面的电荷传递速率; 另一方面部分Cu原子进入Ni体相, 与表层NiO薄膜的形成协同优化了HER中邻近Ni原子的吸-脱附平衡, 显著提升了镍基材料的本征析氢活性. 在催化剂结构特性、 金属协同效应以及表面异质结的协同作用下, NiCu/NF电极表现出了优于大多数非贵金属催化剂的析氢活性及催化稳定性. 在 1.0 mol/L KOH中获得10 mA/cm2的电流密度时, 其需要的析氢过电位仅为86 mV. 动力学分析发现, NiCu/NF电极在析氢过程中遵循Volmer-Heyrovsky机理, 反应速率由电极表面吸附氢原子的电化学脱附过程决定. 镍铜电催化剂纳米立方体结构的构筑为高效析氢电催化剂的设计提供了新的视角, 也可为镍铜基电极材料在能源转化领域的应用发展提供参考.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20200082.
猜你喜欢
粉末冶金技术(2021年1期)2021-03-29 02:34:48
制造技术与机床(2019年6期)2019-06-25 10:17:56
电镀与环保(2018年2期)2018-04-19 02:05:10
电镀与环保(2017年5期)2017-12-19 12:06:09
电镀与环保(2017年2期)2017-05-17 03:42:21
电镀与环保(2016年3期)2017-01-20 08:15:32
电镀与环保(2016年3期)2017-01-20 08:15:28
电镀与环保(2016年2期)2017-01-20 08:15:23
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
