
高效液相色谱-二极管阵列检测器法测定马铃薯及其制品中的α-茄碱和α-卡茄碱
2020-07-06 13:26任兴权刘盼苏菊王蓉曾文锦周丽史蓉李婷婷段亭
食品与发酵工业 2020年12期
任兴权,刘盼,苏菊,王蓉,曾文锦,周丽,史蓉,李婷婷,段亭
(酒泉市食品检验检测中心,甘肃 酒泉, 735000)
马铃薯是全世界重要的粮食作物之一,由于光照、贮藏不当,致使马铃薯发芽或皮变绿,在这一过程中产生了一类甾体糖苷类生物碱[1],被称为龙葵素,其成分为α-茄碱、β-茄碱、γ-茄碱、α-卡茄碱、β-卡茄碱、γ-卡茄碱6种成分,主要以α-茄碱、α-卡茄碱的形式存在,占总的糖苷生物碱的95%。这类生物碱有较强的毒性[2-12],当成年人一次摄入龙葵素超过200 mg时就会引起中毒[2],含量更高时就会导致舌头发麻、呕吐、腹泻、头晕目眩,甚至昏迷、抽搐、呼吸困难、死亡。科学准确地检测马铃薯中α-茄碱、α-卡茄碱的含量对保障我们每一个人的身体健康具有重要的实际意义。
目前已报道的马铃薯中α-茄碱、α-卡茄碱检测方法主要有比色法[13]、液相色谱法[14-25]、液相色谱-质谱法[26-30],后两种方法研究的比较多。邵慧凯等[15]用异丁醇萃取-高效液相色谱法测定马铃薯中的α-茄碱,运用异丁醇萃取,在210 nm波长处检测,从检测谱图可以看出其检测限偏高,达不到实际检测的需求。赵丹青等[16]用V(乙醇)∶V(乙酸)=100∶30溶液提取,过SPE NH2小柱,V(甲醇)∶V(二氯甲烷)=5∶95洗脱用液相检测,只检测了α-茄碱,没有完全将α-茄碱和α-卡茄碱分开,不能准确地测定α-茄碱和α-卡茄碱的各自含量。张舵[21]用无水乙醇-乙酸提取后液相色谱检测,检测的峰型不好,并且有双头峰。王建凤等[26]用V(1%甲酸)∶V(甲醇)=1∶1提取,采用液质法检测。刘红河等[27]用超高液相色谱串联质谱法检测马铃薯及制品中α-茄碱,用V(甲酸)∶V(甲醇)=1∶1作为溶剂提取,并且用MCX固相萃取柱萃取过程复杂,提取效果不是很理想。戴超等[29]运用液质联用法分析贮藏对马铃薯中α-茄碱含量的影响中用V(乙酸)∶V(乙醇)=1∶10混合溶剂振荡提取,其提取效果不佳。现有的液相色谱普遍存在前处理复杂,提取效果差,检出限偏高,检测结果不理想的问题,而现有的液相-质谱联用法则存在提取不完全,对仪器性能要求高等问题。
本研究在综合分析现有方法的基础上,创新了前处理方法,采用液相色谱法分析检测,同时与液相色谱-质谱联用法进行分析比对,该方法操作简单、检测灵敏度高、稳定性好,能有效监测马铃薯龙葵素的含量,为防止食物中毒提供技术支撑,尤其是可以普及应用到基层的检测机构。
1 材料与方法
1.1 材料与试剂
马铃薯,市购。
甲醇、乙腈(均为色谱纯),默克股份两合公司;甲酸(色谱纯),天津市科密欧化学试剂有限公司;无水硫酸钠、无水乙酸镁、无水硫酸镁、无水乙酸钠、乙酸、乙醇、异丁醇(分析纯),天津市科密欧化学试剂有限公司;磷酸二氢钾(色谱纯),天津市科密欧化学试剂有限公司;α-茄碱(C45H73NO15,CAS号:20562-02-1,纯度≥99.9%),多伦多研究化学公司;α-卡茄碱(C45H73NO14,CAS号:20562-03-2,纯度≥99.9%),ChromaDex公司。
1.2 仪器与设备
岛津LC-20AT型液相色谱仪,日本岛津公司;艾柯Advanced-Ⅱ-12型高纯水机,成都唐氏康宁科技发展有限公司;宁波新芝SB25-12DTD型超声波清洗机,宁波新芝生物科技有限公司;长沙英泰TG16型离心机,长沙英泰仪器有限公司;IKA MS3型涡旋混合器,IKA公司;赛多利斯SQP型电子天平,赛多利斯科学仪器(北京)有限公司;梅特勒PL602E/02便携式天平,梅特勒-托利多仪器(上海)有限公司。
1.3 实验方法
1.3.1 HPLC条件
色谱柱:Hubble C18液相色谱柱(250 mm×4.6 mm,5 μm);流动相A为0.02 mol/L磷酸二氢钾溶液,流动相B为乙腈,流动相比例:A为70%,B为30%,流速0.2 mL/min,柱温30 ℃,进样量10 μL,二级管阵列检测器检测范围190~810nm,检测波长195 nm。
1.3.2 标准曲线溶液的配制
分别称取10 mg α-茄碱和α-卡茄碱于10 mL容量瓶中,用甲醇溶解并定容,配制成1mg/mL的标准储备液;用甲醇逐级稀释成0.2、2、10、20、50、100、200 μg/mL的混合标准系列溶液。
1.3.3 样品前处理
用均质器将样品充分打碎混匀,放入分装容器中,密封并标记,于-20 ℃以下冷冻存放。
称取试样1~2 g(精确至0.001 g)于50 mL具塞离心管中,加入3.0~5.0 mL水涡旋混匀1 min,再准确加入25.0 mL酸化甲醇[V(甲酸)∶V(甲醇)=1∶99],涡旋混匀1 min。之后,向离心管中加入1.0~2.0 g 无水硫酸钠,1.0 g 无水乙酸镁。手动剧烈摇动1 min后涡旋混匀1 min。再将离心管置于离心机中,在室温条件下以10 000 r/min的速度,离心5 min。试样为芽和绿皮时取1 mL上清液于10 mL容量瓶中,加入甲醇水溶液[40%(体积分数)甲醇]定容至刻度;试样为马铃薯肉和制品时取5 mL上清液于试管中氮吹浓缩近干,然后加入1 mL甲醇水溶液[40%(体积分数)甲醇]复溶;过微孔滤膜(0.22 μm,尼龙)后上机测定。
1.3.4 定性、定量方法
在以α-茄碱和α-卡茄碱保留时间定性的基础上,与全扫描光谱定性和色谱峰纯度分析相结合的多重定性方法。以外标法对α-茄碱和α-卡茄碱进行定量分析。
2 结果与分析
2.1 提取方式的选择
马铃薯中α-茄碱、α-卡茄碱的常用提取方法有乙醇法、乙醇-乙酸混合剂法、异丁醇萃取法、乙酸-乙腈法、乙醇-乙腈法等,虽然均能不同程度的提取,但是提取效果均不能达到最佳理想状态。本研究先加入少量水再用酸化甲醇提取,通过加入无水硫酸钠、无水乙酸镁这2种在提取中不参加反应而吸收多余的水,最后在高速离心机上离心。由表1可以看出,通过不同方法的对比得出酸化甲醇能够最大限度地提取α-茄碱、α-卡茄碱,同时与吸水性较弱的无水硫酸镁、无水乙酸钠对比,无水硫酸钠和无水乙酸镁的吸水性更强。这是由于在酸性、含水量少的环境中α-茄碱、α-卡茄碱易溶于甲醇,无水硫酸镁、无水乙酸钠有很强的的吸水能力,致使大量的水分被吸收。
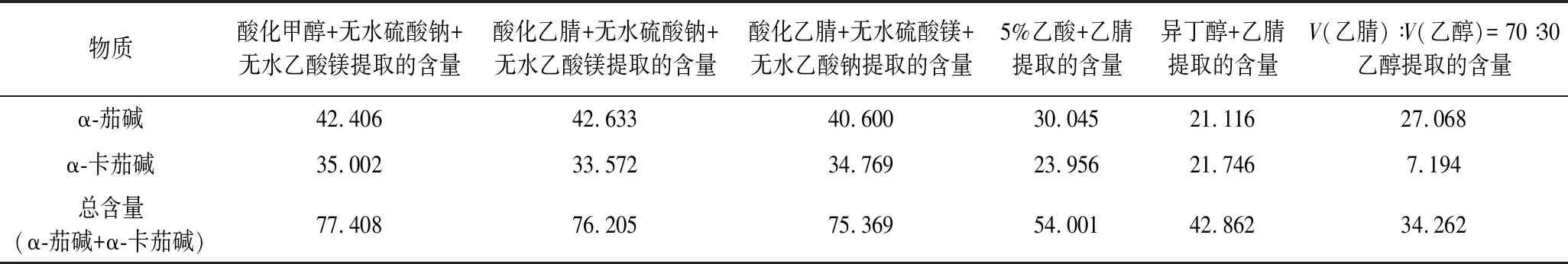
表1 不同试剂提取效果的对比 单位:μg/mL
2.2 色谱条件的选择
2.2.1 检测波长的选择
马铃薯淀粉含量高,在酸性介质中样品易水解,检测基质复杂,同时α-茄碱和α-卡茄碱保留时间相近,仅依据保留时间定性易出现假阳性。二极管阵列检测可进行全波长扫面,通过对待测样品谱图与α-茄碱和α-卡茄碱标准谱图的扫描光谱和最大吸收波长进行比较,同时可进行色谱峰的纯度分析,利用多重定性方法,排除了基质的干扰,防止假阳性结果的出现,提高了定性分析的准确度。
采用二极管阵列检测器的全波长3D扫描功能,在190~810 nm对α-茄碱和α-卡茄碱进行扫描。扫描结果表明α-茄碱和α-卡茄碱的全波长扫描图基本一致,在190~205 nm时扫描图差别较小,在特征波长195 nm处α-茄碱和α-卡茄碱均有最强的紫外吸收,峰面积和峰高度均达到最高值,具体对比结果见图1,因此本研究确定195 nm作为本方法的定性检测波长。
2.2.2 色谱柱的选择
液相色谱法最常用的多为反相色谱C18柱,因为α-茄碱和α-卡茄碱为疏水性物质,并且在提取介质中加入了甲酸,致使提取液的酸性较强,一般的C18柱无法有效分离,本方法采用了OmniBond HPLC Column Hubble C18(250 mm×4.6 mm,5 μm),它采用了超纯硅胶和深度键合这2种技术,具有高柱效,高选择性,高pH稳定性。同时对同类型Hubble C18(150 mm×4.6 mm,5μm)进行对比试验发现无法分离α-茄碱和α-卡茄碱。结果表明HubbleC18(250 mm×4.6 mm,5 μm)色谱柱能够很好的分离、检测α-茄碱和α-卡茄碱,色谱峰的基线平稳。
2.2.3 流动相体系的选择
检测α-茄碱和α-卡茄碱的流动相体系一般由0.4%磷酸/乙腈、0.1%甲酸水溶液/乙腈、甲醇/甲酸铵、甲醇/乙酸铵、甲醇/甲酸、乙腈/磷酸二氢钾。通过比较乙腈体系的基线噪音比甲醇体系小,分离效果更佳,同时,磷酸二氢钾比乙酸铵、甲酸铵、磷酸、甲酸有更好的分离效果。因此,选用乙腈/磷酸二氢钾作为流动相的体系,并且对磷酸二氢钾的浓度和乙腈的配比进行了对比,发现0.2 mol/L磷酸二氢钾能够很好的调节流动相体系的pH,而当V(磷酸二氢钾)∶V(乙腈)=70∶30时流动相体系的pH为6.0,α-茄碱和α-卡茄碱可以完全分开,并且峰型良好,是最佳的分析条件。

a-190 nm波长的谱图;b-195 nm波长的谱图;c-200 nm波长的谱图;d-190、195、200 nm波长下色谱峰的相关信息
图1 不同波长时α-茄碱和α-卡茄碱的色谱图及峰表
Fig.1 Chromatogram and peak table of α-solanine and α-chaconine at different wavelengths
2.2.4 流动相淋洗方式的选择
流动相采用梯度淋洗时能够较早的出峰,但是基线不平稳,致使α-卡茄碱分离的效果不佳。因为α-茄碱和α-卡茄碱出峰时间相差很小,用等度淋洗时,可以保持流动相体系的酸性环境稳定,在有效分离α-茄碱的同时也可有效分离α-卡茄碱。
2.2.5 流动相流速的选择
分别对0.10、0.15、0.20、0.25、0.30、0.40、0.50、0.60、0.80、1.00 mL/min时的分离效果进行了比对。研究结果表明,随着流速的增大,保留时间缩短,峰宽变窄,峰面积降低,α-茄碱和α-卡茄碱的峰重叠越严重。综合保留时间、峰面积、分离度等因素,最终确定流动相流速为0.2 mL/min,。在此优化条件下,α-茄碱和α-卡茄碱分离效果良好,标准谱图见图2,马铃薯芽样品中的α-茄碱和α-卡茄碱谱图见图3。
图2 100.0 μg/mL α-茄碱和α-卡茄碱标准溶液液相色谱图
Fig.2 Liquid chromatogram of 100.0 μg/mL α - solanine and α-chaconine standard solution
图3 马铃薯芽中α-茄碱和α-卡茄碱的色谱图
Fig.3 Chromatogram of α - solanine and α-chaconine in potato bud
2.3 标准曲线与检出限
分别以质量浓度(X, μg/mL)为横坐标、峰面积(Y)为纵坐标绘制标准曲线,α-茄碱和α-卡茄碱的线性方程及其相关系数见表2。由表2可以看出在质量浓度0.20~200 μg/mL时,2种茄碱的线性关系良好,相关系数(R2)为0.999 3~0.999 8,满足定量分析的要求。

表2 α-茄碱和α-卡茄碱的线性方程、相关系数(R2)、 及其方法检出限Table 2 Linear equation, correlation coefficient (R2) and detection limit of α-solanine and α-chaconine
以不含茄碱的新鲜马铃薯肉作为空白基质,在其中加入低浓度α-茄碱和α-卡茄碱,按照本研究的上述方法进行前处理和分析检测,以3倍、10倍信噪比分别计算得到本方法中α-茄碱和α-卡茄碱的检出限均为0.5 mg/kg,定量限均为1.7 mg/kg,说明本方法的灵敏度很高。
2.4 回收率和精密度
选用已测定α-茄碱和α-卡茄碱含量的马铃薯芽,分别添加4.0、6.0、10.0 μg/mL 三个质量浓度水平的α-茄碱和α-卡茄碱混合标准溶液,配成加标样品,按照上述方法进行前处理,每个加标水平的样品平行测定6次,结果加标回收率为98%~107%,相对标准偏差为0.05%~0.44%(表3),说明本方法有较高的准确度和精密度,完全适合马铃薯中α-茄碱和α-卡茄碱的分析检测。
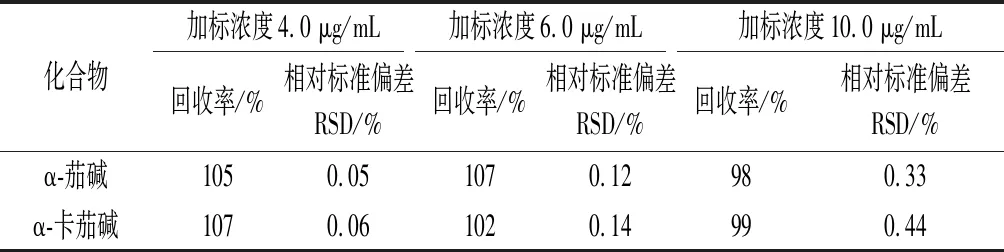
表3 α-茄碱和α-卡茄碱的回收率和精密度Table 3 Recoveries and RSDs of α-solanine and α-chaconine
2.5 马铃薯样品分析
用本方法对马铃薯的芽、绿皮、肉、马铃薯淀粉、马铃薯粉条的5种样品平行测定2次,平均结果见图4,由检测结果可看出,马铃薯芽中的α-茄碱和α-卡茄碱含量是很高的,在马铃薯芽、绿皮、肉、淀粉、粉条依次降低。
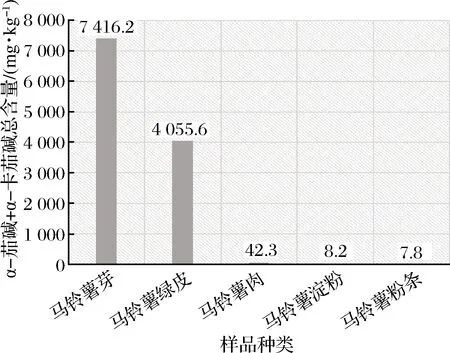
图4 马铃薯样品中龙葵素含量(α-茄碱+α-卡茄碱) 柱形图
Fig.4 Column chart of α-solanine and α-chaconine content in potato samples
2.6 与液-质法的对比
对马铃薯的肉、白芽、绿芽分别用本研究方法和土豆及其制品中α-茄碱和α-卡茄碱的测定补充方法中液相色谱-串联质谱仪法(BJS201806)进行提取、测定,检测结果见表4,可以看出2种方法的检测结果的相对偏差为4.0%~8.8%,本研究的液相色谱法检测的含量均比补充方法BJS201806高。说明本方法能够更加科学准确地测定α-茄碱和α-卡茄碱的含量。
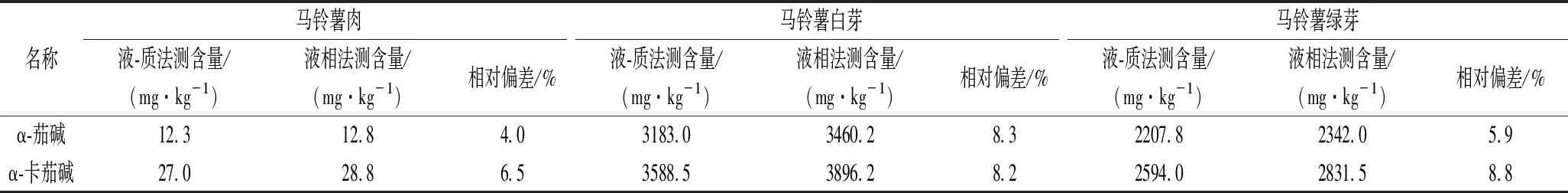
表4 液相色谱法与液相-质谱法检测结果的对比Table 4 Comparison between the results of liquid chromatography and liquid mass spectrometry
3 结论
本研究建立了高效液相色谱-二极管阵列检测器测定马铃薯中α-茄碱和α-卡茄碱的方法。利用酸化甲醇提取,以二极管阵列检测器的全扫描和色谱纯度分析进行定性,很大程度解决了现有报道文献方法中提取不完全、分析检测不准确的问题。该方法前处理简单、灵敏度高,具有良好的精密度、准确度,可作为马铃薯及其制品中α-茄碱和α-卡茄碱监测分析,防止马铃薯存储不善而食用后引起的食物中毒事件发生,该方法可普及到市、县级基层检验检测机构,对于食品安全监管具有重要的现实意义。
猜你喜欢
煤化工(2022年3期)2022-07-08
杭州电子科技大学学报(自然科学版)(2022年3期)2022-06-08
当代水产(2022年4期)2022-06-05
口腔护理用品工业(2021年4期)2021-11-02
阅读(科学探秘)(2021年8期)2021-09-01
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
首都食品与医药(2020年1期)2020-10-21
中国科学院大学学报(2019年1期)2019-01-21
山东工业技术(2016年10期)2016-09-06
