
产絮凝剂菌株Pseudoalteromonas undina JP 的表型特征及基因组学分析
2020-06-19 17:04:06王玉霞周海军
浙江海洋大学学报(自然科学版) 2020年5期
王玉霞,崔 霞,周海军,穆 军
(浙江海洋大学海洋科学与技术学院,浙江舟山 316022)
生物絮凝剂因其绿色生态,无毒无害,活性高,不会引入二次污染,且絮凝的范围广,与可能会引起多种环境和人类健康问题相比的无机絮凝剂和有机合成絮凝剂相比,生物絮凝剂被认为是一种环境友好型的高效絮凝剂,在污水、废水的处理和水质的净化中具有很好的发展前景[1-5]。菲律宾蛤仔Ruditapes philippinarum 作为世界上最受欢迎的海洋双壳类水产养殖动物之一,其产生的黏附污泥是一种典型的水产养殖副产品废物。来自中国舟山水产养殖场的菲律宾蛤仔黏附污泥(R.philippinarum conglutination mud,RPM)被报道为一种具有前景的天然生物絮凝剂资源,研究发现RPM 中含有有效絮凝多糖,其共附生微生物可能是絮凝多糖的产生源。RPM 对不同染料溶液的脱色率均在90%以上,最大絮凝率高达91.8%[6-7]。从RPM 中分离得到的菌株最大絮凝率可达88.14%,最大脱色率可达90.02%[7-8]。
本研究从RPM 中分离筛选得到一株具有高絮凝活性的菌株JP,通过16S rDNA 测序比对、系统发育树构建以及生理生化特征对其鉴定到种。然后通过Illumina 测序平台对菌株JP 的全基因组框架进行扫描测序,将测得序列进行基因预测并注释到COG、GO、KEGG、NR 和Swiss-Prot 数据库,通过对其代谢功能的分析来揭示其产絮凝剂的分子机制。
1 材料和方法
1.1 产絮凝剂菌株JP 的来源
从具有高絮凝活性的菲律宾蛤仔黏附污泥中分离、筛选、纯培养获得的菌株。
1.2 絮凝率测定
菌株的絮凝率参照MU Jun,et al[6]使用的高岭土悬浮液法进行测定。配制4 g·L-1的高岭土悬浮液93 mL,分别加入5 mL 10 g·L-1氯化钙溶液和2 mL 待测菌株JP 的菌悬液,调节pH 至7.5。200 r·min-1搅拌混合液1 min,然后以80 r·min-1搅拌2 min。静置10 min 后,取液面下1 cm 处悬浊液于便携式可见分光光度计在550 nm 吸光度下进行光密度值(OD)测定。空白样品以2 mL 去离子水代替待测样品进行测定,絮凝率(flocculation rate,FR)计算公式为:
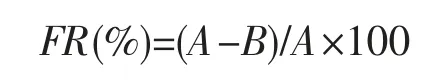
A 和B 分别为空白对照组和待测样品组的OD550值。
1.3 生理生化性状测定
菌株JP 的形态特征、生理生化指标、生长温度与盐度范围、胞外酶活性检测、碳源的利用测定方法参照《伯杰氏细菌鉴定手册》[9]、《常见细菌系统鉴定手册》[10]以及BAUMANN,et al[11]。
1.4 16S rDNA 测序
将菌株JP 接种到液体培养基中恒温25 ℃培养10 h 后使用TaKaRa 全基因组提取试剂盒(DNA Extraction Kit Ver.3.0)进行核酸DNA 的提取和纯化。利用引物27F(5’-AGA GTTTGATCATGG CTC AG-3’)和1492R(5’-TAC GGTTACCTGTTACGACTT-3’)对菌株抽提的核酸进行PCR 扩增[12],反应体系终体积为20 μL,包含2.0 μL 10×Ex Taq buffer、1.6 μL 2.5 mmol·L-1dNTP mix、5p 引物27F、5p 引物1492F、0.5 μL 模板、0.2 μL Ex Taq 和14.1 μL ddH2O。扩增反应程序如下:95 ℃,5 min;95 ℃,30 s,55 ℃,30 s,72 ℃,90 s,24个循环;72 ℃,10 min。扩增结果以凝胶电泳进行检测,检测合格后使用ABI 3730XL 测序仪进行测序,将测序后的序列进行拼接。
1.5 分子鉴定及系统发育树构建
将拼接后的序列于EzBioCloud(http://www.ezbiocloud.net)进行blast 比对,将菌株鉴定到种属水平。菌株JP 的16S rDNA 序列上传到NCBI 数据库,登录号为KX702268。菌株JP 于2016 年8 月29 日保藏于中国微生物菌种保藏管理委员会普通微生物中心,保藏号为CGMCC No.12914。选取相似比对结果中相似度高的模式菌株及外间组模式菌株通过软件MEGA7 进行系统发育树的构建,分析其进化关系。
1.6 菌株JP 的全基因组框架扫描
将菌株JP 纯培养后提取基因组DNA,利用1%琼脂凝胶电泳检测并收集抽提的DNA。对质检收集后的DNA 进行300~500 bp 片段化,然后使用TruSeqTMDNA Sample Prep Kit 进行文库构建,TruSeq PE Cluster Kit 进行桥式PCR,最后使用TruSeq SBS Kit 利用Illumina 测序技术对DNA 进行paired-end(PE)测序。将测序得到的原始数据进行有效数据的统计、序列拼接(SOAPdenovo v2.04 软件)、序列矫正(GapCloser v1.12 软件),得到拼接后的序列,以便进行下一步分析。
1.7 生物信息学分析
对拼接后的序列进行rRNA/tRNA 的查找(RNAmmer-1.2 和tRNAscan-SE v1.3.1 软件)、串联重复序列预测(http://tandem.bu.edu/trf/trf.basic.submit.html)[13]、插入序列预测(https://www-is.biotoul.fr/)[14]、基因的功能预测(Glimmer 3.02 软件),并将预测基因的蛋白序列与Nr、genes、string 和GO 数据库进行blastp 比对(BLAST 2.2.28+软件),得到预测基因的功能注释信息。
2 结果与讨论
2.1 絮凝率、16S 序列比对结果及进化关系分析
分离纯化的单菌株JP 恒温25 ℃纯培养10 h,测得其絮凝率为80.0%。比对结果显示菌株JP 的16S序列与Pseudoalteromonas undina NCIMB 2128T 的序列具有100%相似性。菌株JP 的系统进化发育树如图1,低于80%的值被隐藏,由图1 可知,菌株JP 与P.undina 聚集在一个分支。
菌株的生理生化指标测定结果见表1。由表1 可知,菌株JP 为杆状菌,革兰氏阴性菌,具有极性鞭毛;生长盐度耐受范围与模式菌株相比,由1%~12%提升到了15%;高温不耐受,37 ℃无法生长;能产生酶有淀粉酶、几丁质酶、精氨酸和酪氨酸;能利用葡萄糖、麦芽糖,而阿拉伯糖、半乳糖、蜜二糖、乳糖、甘露醇、山梨糖醇、柠檬酸盐、木糖均无法利用。与模式菌株的理化特征比对,结合16S 序列比对和系统发育树结果,菌株JP 被鉴定为P.undina。
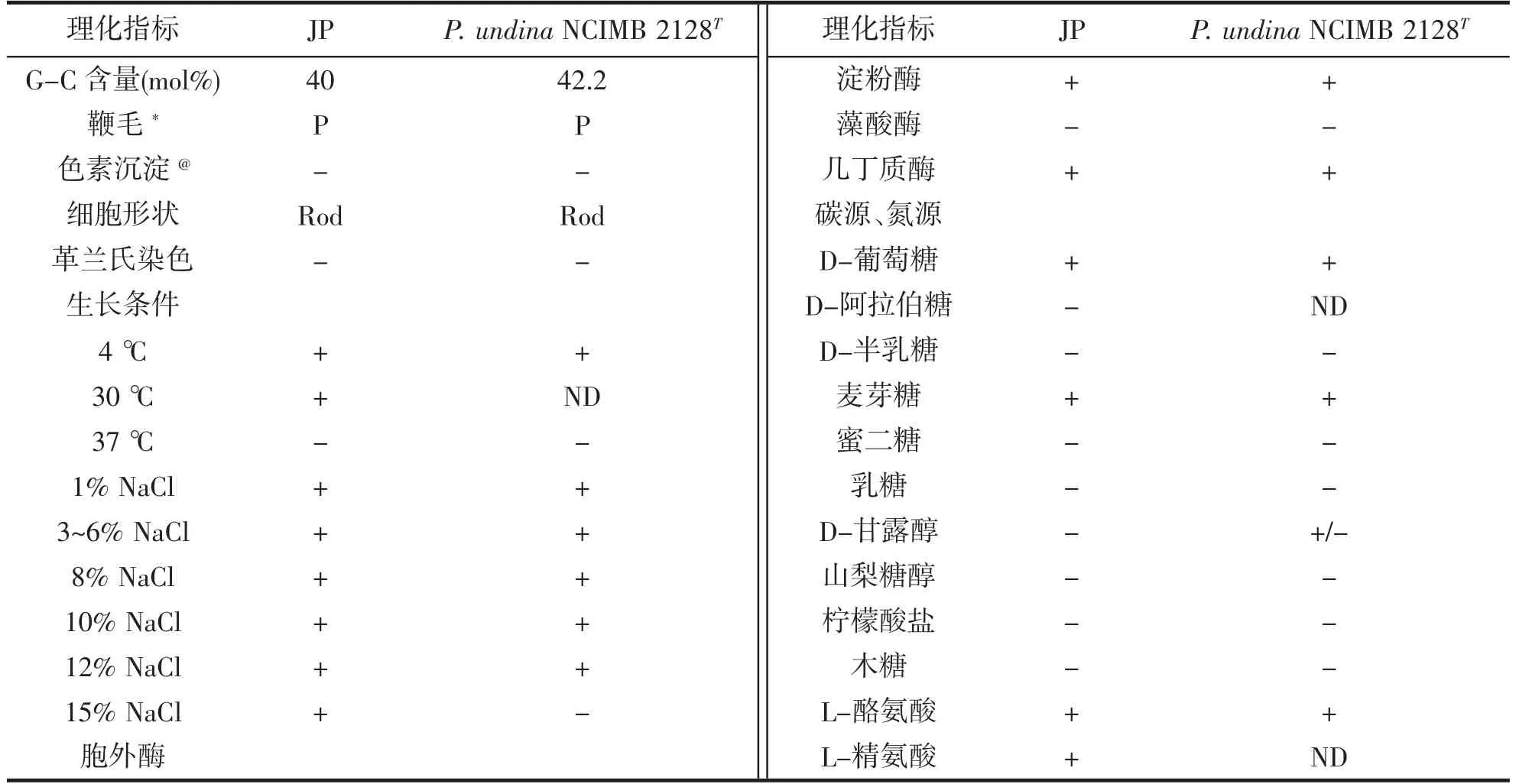
表1 菌株JP 的生理生化特征测定结果Tab.1 The results of biochemical tests of strains JP
2.2 菌株JP 的基因组基本特征
为了深入研究RPM 共附生微生物-产絮凝剂微生物的产絮凝剂分子机制,本研究利用Illumina 技术对菌株JP 的基因组进行了框架扫描测序,序列拼接为20 个大框架序列,总长度为4 046 116 bp,其中18 个框架序列长度大于1 000 bp,最长框架为885 303 bp,框架N50 为480 011 bp,N90 为113 970 bp,GC 含量为40。将菌株JP 拼接后的序列进行与基因预测,包含14 个rRNA、87 个tRNA 和3 746 个基因。
2.3 重复序列预测
通过TRF(tandem repeat finder)对菌株JP 的基因组进行串联重复序列的在线预测,共预测到111 条串联重复序列,44.1%的序列匹配分值为50~100,37.8%的序列匹配分值为101~500,其余匹配分值大于500,表明菌株JP 的基因组中多存在短串联重复序列。通过IS Finder 对菌株JP 的插入序列进行在线预测,24条潜在的插入序列被筛选出,筛选结果以E-value(0.001)为标准,IS3 序列家族占绝对优势,仅1 条属于IS66 家族。
图2 菌株JP 的基因组CG 图谱Fig.2 The CG view of P.undina JP
2.4 COG 功能分析
将注释基因与COG 数据库进行比对,结果表明2416 个(64.5%)基因具有COG 编号,即具有明确的生物学功能(图3)。其中,COG 功能一级分类中参与代谢的蛋白数最多,高达1 004 个(41.6%),其次是参与细胞内进程和信号传导功能(33.4%),以及信息储存和处理(21.4%)。COG 功能二级分类中,拥有最多功能基因的是类别J(翻译、核糖体结构和生物发生,9.8%)、E(氨基酸转运和代谢,9.6%)、M (细胞壁/膜/包膜生物发生,7.4%)和T(信号转导机制,7.3%)。类别Q 的功能基因仅占2.2%(次生代谢产物的生物合成、转运和分解代谢)。

图3 菌株JP 的COG 的功能分类Fig.3 The results of COG second level class in P.undina JP
2.5 GO 功能分析
GO 功能注释结果统计如图4 所示。由图可知,61.3%(2 295)的预测基因具有GO 注释信息。其中,在生物学过程中,细胞内过程、单生物过程和代谢过程占优势;在细胞组分中,细胞部位、细胞膜部位、细胞膜和细胞是优势功能组;在分子功能中,结合和催化活性占绝对优势。菌株JP 是一株具有絮凝活性的功能性菌株,其生长繁殖代谢是重要的生命活动,其中需要大量的相关代谢、生物调控以及代谢产物的运输等基因表达,因此可以合理地解释菌株JP 的这些功能基因表达占优势地位。
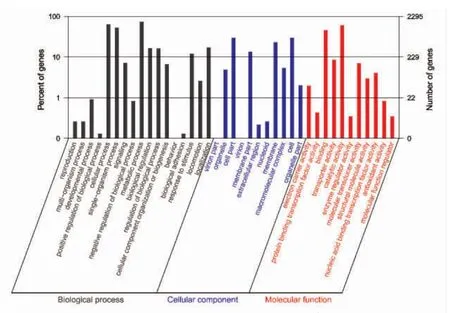
图4 菌株JP 的GO 功能分类图Fig.4 Functional classification of GO annotation in P.undina JP
2.6 KEGG 代谢通路分析
将菌株JP 基因组预测的基因序列与KEGG 数据库进行比对,结果显示在菌株JP 基因组中预测的3 746 个全长基因中,有2 094 个基因(55.9%)匹配到KEGG 数据库中的基因,且在其对应的代谢途径上均进行了定位,共参与了193 条KEGG 的代谢途径。其中参与代谢途径的总共535 个基因,参与次生代谢产物的生物合成有240 个基因,参与不同环境的微生物代谢共有165 个基因。此外,参与碳代谢的有86 个基因,主要包含肽聚糖、脂多糖、氨基糖、核苷酸糖、淀粉、蔗糖、果糖、甘露糖的生物合成与代谢。迄今为止,对于细菌产絮凝剂机制的报道众说纷纭,但其明确的絮凝基因等仍处于探索中,但是目前已有报道一些胞外絮凝剂多糖的生物合成路径及关键前体的合成,如纤维素和普鲁兰多糖的合成需要先前体UDP-D-Glucose,黄原胶合成的前体为UDP-Glucose、GDP-mannose 和UDP-glucuronate,可得然胶需要先合成前体UDP-Glucose。本研究中能明确找到这些合成前体,然而关于目前已经明确的eps 合成基因簇并未在注释的代谢通路中寻找到,但能发现一些胞外多糖合成酶的关键调控基因,如纤维素、海藻酸钠合成最开始需要c-di-GMP 与PilZ 的结构域结合来激活[16-18],铜绿假单胞菌中胞外多糖的合成受AlgR 等基因的调节[19]。也发现了一些目前所报道的胞外多糖聚合前的重复单元合成,如UDP-ManNAcA、dNTP-D-Glu、UDP-3-O-(3-acetyl)-GlcNAc 等,这些重复单元在相应的聚合酶作用下聚合成多糖,多糖通过转运系统等分泌到胞外,形成胞外多糖絮凝剂,使微生物具有絮凝活性[20-23]。
暂未预测到目前已报道的胞外多糖合成基因簇,有多种原因导致此结果:一是目前关于P.undina产絮凝剂这一特征是本实验室首次研究报道的功能菌株,国内外研究者对于该菌种的研究呈现明显的不足;二是由于考虑到研究原因,并未对菌株JP 进行完整的基因组测序,仅对框架进行扫描,框架之间断开的序列有可能具有功能基因,因此造成数据的部分缺失;三是目前关于微生物产絮凝剂的合成路线仍处于探索阶段,其中的原理与机制大量缺乏,因此造成本研究菌株中预测不到胞外多糖的合成基因簇;四是该菌株具有类似eps 基因簇的基因所合成的产物承担该功能。
2.7 NR 功能分析
将菌株JP 组装后预测的基因注释到NR 数据库,97.5%(3 653)的预测基因在NR数据库具有匹配信息。结果表明24.6%的E 值位于1×10-180~1×10-100,13.7%位于1×10-100~1×10-50,3.9%位于1×10-50~1×10-30,4.1%位于1×10-30~1×10-5;注释信息表明79.0%的基因序列具有100%的相似性,20.0%的相似性集中在80%~99%,其中97.8%的预测基因与Pseudoalteromonas sp.有相似的同源序列,其中35.2%(相对于注释到Pseudoalteromonas sp.中相似基因的百分比,下同)的预测基因与P.undina 相似,20.2%的预测基因与P.tetraodonis相似。菌株JP 与P.undina 同属一个物种,且与其余3 个物种分类学地位相近因此大部分基因功能相似是合理的。
2.8 Swiss-Prot 数据库注释
将菌株JP 的预测基因注释到Swiss-Prot 数据库,2 540 个基因在该数据库有注释信息,占总基因数的67.8%。对E 值分布进行统计,5.9%基因的E 值位于1×10-10~1×10-5,24.1%位于1×10-30~1×10-5,13.5%位于1×10-50~1×10-30,24.1%位于1×10-100~1×10-50,19.8%位于1×10-180~1×10-100。1.1%的基因序列具有100%的相似性,26.6%的相似性集中在80%~99%,35.3%相似性为60%~79%,24.6%相似性为50%~59%。
2.9 次级代谢产物合成基因簇
通过antiSMASH 对菌株JP 的序列进行次级代谢产物合成基因簇预测,分别在JP_scaffold2 和JP_scaffold4 序列中各预测到一个基因簇,分别为铁载体(siderophore)和细菌素(bacteriocin)合成基因簇。
3 结论
从RPM 中分离筛选的高絮凝活性菌株JP 通过16S 测序和生理生化指标测定鉴定为P.undina,絮凝活性达到80.0%。通过一系列的预测对菌株JP 的基因组进行了初步分析,一共预测到了87 个tRNA,14个rRNA,111 条串联重复序列,24 条插入序列,3 746 个基因和2 个次级代谢产物合成基因簇。将预测的基因进行数据库的注释,64.5%、61.3%、55.9%、97.5%、67.8%的基因分别在数据库COG、GO、KEGG、NR 和Swiss-Prot 匹配到了相应的注释信息。尽管没有注释到一个完整的与目前所报道的胞外多糖代谢路径一致的,这也有可能是目前这一类研究领域数据大量缺乏的原因,如果需要进一步探索菌株的代谢通路,未来还需要更深入的分子水平研究。
猜你喜欢
今日农业(2021年11期)2021-08-13 08:53:24
西北农林科技大学学报(自然科学版)(2019年8期)2019-07-17 02:43:32
钻井液与完井液(2018年5期)2018-02-13 01:07:26
华东纸业(2016年2期)2017-01-19 07:37:32
中国塑料(2016年10期)2016-06-27 06:35:36
遗传(2015年5期)2015-02-04 03:06:55
海洋科学(2014年12期)2014-12-15 03:35:00
应用化工(2014年10期)2014-08-16 13:11:29
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
