
CITED2 and the modulation of the hypoxic response in cancer
2020-06-19 06:34nicaFernandesSofiaCaladoLeonardoMendesSilvaJosBragan
Mónica T Fernandes, Sofia M Calado, Leonardo Mendes-Silva, José Bragança
Abstract CITED2 (CBP/p300-interacting transactivator with Glu/Asp-rich C-terminal domain, 2) is a ubiquitously expressed protein exhibiting a high affinity for the CH1 domain of the transcriptional co-activators CBP/p300, for which it competes with hypoxia-inducible factors (HIFs). CITED2 is particularly efficient in the inhibition of HIF-1α-dependent transcription in different contexts, ranging from organ development and metabolic homeostasis to tissue regeneration and immunity, being also potentially involved in various other physiological processes. In addition, CITED2 plays an important role in inhibiting HIF in some diseases, including kidney and heart diseases and type 2-diabetes. In the particular case of cancer, CITED2 either functions by promoting or suppressing cancer development depending on the context and type of tumors. For instance,CITED2 overexpression promotes breast and prostate cancers, as well as acute myeloid leukemia, while its expression is downregulated to sustain colorectal cancer and hepatocellular carcinoma. In addition, the role of CITED2 in the maintenance of cancer stem cells reveals its potential as a target in non-small cell lung carcinoma and acute myeloid leukemia, for example. But besides the wide body of evidence linking both CITED2 and HIF signaling to carcinogenesis, little data is available regarding CITED2 role as a negative regulator of HIF-1α specifically in cancer. Therefore, comprehensive studies exploring further the interactions of these two important mediators in cancer-specific models are sorely needed and this can potentially lead to the development of novel targeted therapies.
Key words: Cancer; Cancer stem cell; CBP/p300; CITED2; Hypoxia-inducible factors 1α;Hypoxia; Leukemia; Tumor
INTRODUCTION
CITED2, formerly known as melanocyte-specific gene related gene 1 and p35srj[1-4], is a member of the CBP/p300-interacting transactivator with Glu/Asp-rich C-terminal domain family of transcriptional modulators that includes also CITED1, CITED3 and CITED4. These proteins, characterized by a conserved CBP/p300-interacting domain located at the C-terminal domain, termed CR2 for conserved region 2 (Figure 1A), are only conserved in vertebrates and contribute to several aspects of embryogenesis and/or to normal organ function in adult organisms[1,3,5-7]. CBP and p300 are homologous nuclear phosphoproteins, ubiquitously expressed, that contain amongst others, three highly conserved cysteine–histidine-rich domains (CH1, -2, and -3) and function as transcriptional coactivators[8,9]. Although CITED proteins lack a DNAbinding domain, they usually localize in the nucleus where they function as transcriptional modulators[10,11]. From all CITED members, CITED2 has been the most studied, due to its pivotal roles in many different biological processes[5,6,12-14]. The CR2 of CITED2 is composed of 32 amino acids (residues 224 to 255) that contain a potent transactivation domain (TAD), which is responsible for physically interacting with the CH1 domain of CBP/p300, also known as TAZ1[3](Figure 1A). The CH1 domain of p300, which has an extremely high affinity for CITED2, also binds hypoxia-inducible factors (HIF)-1α[15]and other transcription factors such as the NFκB p65 subunit[16],p53, and MDM2[17]. Due to its interaction with CBP/p300, CITED2 was reported to act both as a positive and a negative regulator of transcription. This function appears to depend on whether CITED2 enables the interaction between CBP/p300 and other transcription factors, or whether it prevents such interactions. CITED2 has been reported to be a transcriptional coactivator of TFAP2, SMAD2/3 (mediating TGFβ signaling), peroxisome proliferator-activated receptors, ISL1, amongst other factors[6,18-20]. Conversely, it acts as a transcriptional repressor through interfering with the binding of transcription factors, such as HIF-1α and STAT2, with CBP/p300[21].CITED2 is ubiquitously expressed and its expression is modulated by many biological stimuli including lipopolysaccharide and cytokines, such as interleukin 9 and interferon gamma, in different cell types[2]. Most notably, CITED2 is highly inducible by the HIFs under low oxygen/hypoxic conditions[3].
CITED2 AND THE MODULATION OF THE HYPOXIC RESPONSE

Figure 1 Structure of CITED2 and its interplay with hypoxia-inducible factors to modulate the response to hypoxia. A: Schematic representation of CITED2,hypoxia-inducible factor (HIF)-1α and CBP/p300. The conserved regions amongst CITED family members, and the serine-glycine rich junction domain unique to CITED2 are indicated. Conserved regions 2 is the domain that characterizes the CITED proteins that contains a potent transactivation domain (TAD), which is responsible for the physical interaction with the domain CH1 of CBP/p300, also represented. HIF-1α has two TADs, namely, a N-terminal TAD and a C-terminal TAD,which are responsible for the transcriptional activity under hypoxia. To this end, HIF-1α binds to the CH1 region of p300, but CITED2 was shown to compete out HIF-1α for the binding to the same region, and to interfere with hypoxia-driven transcription; B: Model for CITED2, HIF-1α and CBP/p300 in hypoxia-responsive gene regulation. In normal levels of oxygen (normoxia), HIF-1α is hydroxylated at two proline residues within its oxygen-dependent degradation domain, which marks HIF-1α for proteasome degradation. In normoxia, CITED2 binds to CBP/p300 in the nucleus regulating the expression of target-gene. In addition, part of CITED2 proteins is ubiquitinated by FBXL5 and degraded by the proteasome. In hypoxia, HIF-1α is no longer hydroxylated, which prevents its degradation by the proteasome.Consequently, HIF-1α translocate and accumulates in the nucleus to associate with the HIFβ subunit to bind to specific Hypoxia-Response Elements in hypoxiaregulated genes. CITED2 competes with HIF-1α for the binding to the CH1 region of CBP/p300 and interferes with hypoxia-driven transcription. CR: Conserved regions; SRJ: Serine-glycine rich junction; TAD: Transactivation domain; HIF: Hypoxia-inducible factor; NAD: N-terminal TAD; CAD: C-terminal TAD.
The cellular response to hypoxia is critical for cell survival and is strictly regulated to allow cells to adjust their needs during altered oxygen levels. HIFs play a central role in systemic and cellular adaptation to decreased oxygen levels. These transcription factors are heterodimeric proteins consisting of a hypoxia-inducible HIFα subunit,and a constitutively expressed HIFβ subunit, also known as Aryl Hydrocarbon Receptor Nuclear Translocator or ARNT[22,23]. Three oxygen-sensitive HIF subunits have been identified to date, HIF-1α, HIF-2α, and HIF-3α, and all of them dimerize with HIF-1β, originating HIF-1, HIF-2, and HIF-3 heterodimers, respectively[24]. HIF-1α, HIF-2α and HIF-3α have a high degree of sequence homology but are significantly distinct in their functions. For instance, the hypoxic response may be exclusively controlled by one or the other oxygen-sensitive HIF isoforms in different contexts[25].Since several studies have reported a role for CITED2 in hypoxia, particularly through the inhibition of the HIF-1α, we will focus the review on this isoform.
HIF-1α cellular levels are essentially regulated through alterations in protein stability. In normal oxygen levels (normoxia), HIF-1α (and also HIF-2α) is constantly expressed, but rapidly degraded through the ubiquitin-proteasome pathway, in an oxygen-dependent manner[26-29]. HIF-1α contains an oxygen-dependent degradation domain (ODDD) which is hydroxylated at two proline residues by prolyl-4-hydroxylase domain proteins (PHDs), known as oxygen sensors[30,31]. The prolylhydroxylation allows the interaction of HIF-1α with the von Hippel-Lindau (VHL)protein, which is the substrate recognition component of an E3 ubiquitin ligase complex, leading to HIF-1α degradation by the proteasome[32-36](Figure 1B). HIF-1α has two TADs, namely, a N-terminal TAD (NAD) located within the ODDD and a Cterminal TAD (CAD), which are responsible for the transcriptional activity of HIF-1α(and HIF-2α) under hypoxia (Figure 1A). In addition to the ODDD regulation, which prevents the stability of HIF proteins, a second oxygen-dependent mechanism is responsible for the inhibition of the HIF-1α transcriptional activity. Indeed, an asparagine residue located in the HIF-1α CAD (and HIF-2α CAD), is the target for hydroxylation by an oxygen-dependent hydroxylase named factor inhibiting HIF or FIH-1[21,37]. The hydroxylation of this asparagine residue interferes with the recruitment of the CBP/p300 in normoxia[38,39]. Therefore, molecular mechanisms involving the hydroxylation of ODDD and CAD synergize to neutralize HIF-1α (and HIF-2α) transcriptional activity in normoxia.
In hypoxia, the prolyl-4-hydroxylase domain proteins lose their ability to hydroxylate HIF-1α, which prevents its interaction with VHL and proteasomal degradation. Consequently, HIF-1α translocates and accumulates in the nucleus, and associates with the HIFβ subunit to form HIF-1 and to bind to specific hypoxiaresponse elements in hypoxia-regulated genes (Figure 2B). Thus, HIF-1 is responsible for transcriptional programs that promote erythropoiesis, glycolysis, and angiogenesis, between other processes[22,40,41]. It was previously demonstrated that HIF TADs play different roles in hypoxia responses by activating distinct subsets of genes[42,43]. Nevertheless, the expression of hypoxia-responsive genes is only efficient when HIF-1 recruits CBP/p300[15].
CITED2 was shown to compete with HIF-1α for the binding to the CH1 region of CBP/p300, and to interfere with hypoxia-driven transcription[3](Figure 1A). Structural studies have shown that both HIF-1α and CITED2 transactivation domains interact with the CH1 domain of CBP/p300 through helical motifs that flank a conserved LP(Q/E)L sequence to achieve high affinity binding, displacing each other in a feedback loop during the hypoxic response[10,43-45]. More specifically, Berlowet al[45]have shown that CITED2 is able to displace HIF-1α by forming a transient ternary complex with TAZ1/CH1 and HIF-1α, competing for a shared binding site through its LPEL motif, resulting in a TAZ1 conformational change that increases the rate of HIF-1α dissociation, even at modest concentrations of CITED2[45]. Moreover, CITED2 is itself an hypoxia-stimulated gene through the action of HIF-1α and HIF-2α[3,46].Thus, CITED2 is thought as a negative feedback regulator of HIF-1α, contributing for the cellular mechanisms that attenuate the hypoxic response.
In addition to reducing the hypoxia-induced activation of HIF CAD-dependent genes, CITED2 also interferes with HIF NAD-dependent gene activation. Indeed,Yoonet al[47]showed that NAD interacts with the CH1 and CH3 domains of p300 and these were both required for NAD-dependent transactivation. Interestingly, CITED2 was shown to be able to inactivate NAD by interfering with the NAD-CH1 binding,but not with the NAD-CH3 interaction[47]. Nevertheless, CITED2 is a powerful inhibitor HIF-1α transcriptional activity by blocking the recruitment of CBP/p300 through both NAD and CAD. Remarkably, the same authors found that NAD activation through binding to p300 could be blocked not only by CITED2 but also by the VHL protein, supporting the hypothesis that NAD might be controlled by VHL protein during normoxia and by CITED2 during hypoxia[47]. In this context, we have reported another mechanism supporting a role for CITED2 in HIF-1α NAD inactivation in normoxia. Indeed, we have demonstrated that F-Box and leucine-rich repeat protein 5 (FBXL5) overexpression in cells, results in CITED2 degradation,which enabled the transcriptional activity of HIF-1α through the NAD in normoxia(Figure 1B). In fact, we showed that CITED2 and FBXL5 proteins interact, and FBXL5,which is a substrate adaptor protein part of E3 ubiquitin ligase complexes, triggers a proteasome-dependent degradation of CITED2 and consequently contributes to the modulation of gene expression mostly through HIF-1α NAD[11].

Figure 2 Roles of CITED2 in homeostasis and cancer. CITED2-dependent modulation of hypoxia-inducible factor (HIF) is fine-tuned to control physiological processes such as development, tissue regeneration and immunity. Abrogation of CITED2/HIF-1α homeostasis may lead to the initiation, and/or disease progression depending on the cellular context. Cancer may be one these diseases, in which CITED2 and HIF-1α intervene to control tumor growth, drug resistance and metastization. HIF: Hypoxia-inducible factor.
CITED2 AND HYPOXIA IN PHYSIOLOGICAL PROCESSES
The role of CITED2 in regulating HIF1 activity was shown to be also essentialin vivo.Indeed,Cited2knockout mouse embryos died in utero at around mid to late gestation and displayed multiple developmental anomalies, such as heart malformations,adrenal agenesis and neural tube defects[6]. Interestingly, the hearts of Cited2-null embryos displayed elevated expression of HIF-1α target genes, such as vascular endothelial growth factor (VEGF), glucose transporter 1, and phosphoglycerate kinase 1. These genes were also upregulated in Cited2-depleted mouse embryonic fibroblasts under hypoxic conditions[48]. Interestingly, a normalized expression of HIF-1α target genes and part of the heart defects were rescued upon the heterozygous deletion of anHif1aallele in Cited2-null embryos[48,49]. This indicated that a hyperactivity of HIF-1α in Cited2-null embryos is, at least in part, responsible for the cardiac developmental anomalies observed. Cited2-deficient mouse embryos present also defects in the developing eye and showed an excessive and disorganized hyaloid vasculature with increased VEGF expression[50]. Further experiments showed that deletion ofHif1ain the lens could specifically rescue the previous phenotype, supporting the notion that Cited2 is required for the proper hyaloid vascular system regression through negative modulation of HIF-1 signaling during eye development[50]. Cited2 was also reported to modulate HIF-dependent expression of VEGF in the nucleus pulposus of the rat intervertebral disc, most likely contributing for the mechanism by which the cells of the nucleus pulposus survive in a hypoxic environment[51].
Normal hematopoiesis also relies on the correct function of CITED2. Indeed,CITED2 is crucial for the maintenance of the hematopoietic stem cell (HSC) pool in the bone marrow (BM), and its conditional deletion in the hematopoietic system resulted in a dramatic loss of adult HSCs and primitive progenitor cells, ultimately leading to BM failure[52]. In addition, it was reported that Cited2-deficient HSCs presented impaired quiescence and reconstitution capacity, which could be partially rescued by additional deletion of HIF-1α[53]. Therefore, Cited2 is able to regulate HSC quiescence through a HIF-1-dependent mechanism, although other HIF-1-independent mechanism(s) may be also involved in this context[53]. In addition, CITED2 was reported to be involved in immunity, more specifically by repressing macrophagemediated inflammation. Thus, Cited2 deficiency may enhance proinflammatory gene expression through stabilization of HIF-1α in macrophages. Furthermore, the inhibition of HIF-1α in Cited2-deficient macrophages completely reversed the elevated proinflammatory cytokine/chemokine gene expression[54]. Thus, the repressive CITED2 action on HIF-1α activity is crucial for many aspects of mouse and rat development, as well as adult homeostasis.
CITED2 AND HYPOXIA IN DISEASE
CITED2 was shown to be involved in the regulation of the hypoxic response in the context of diseases (Figure 2). Using animal models of chronic kidney disease and heart failure, Tanakaet al[55]have implicated CITED2 in the negative regulation of HIF-target genes, and consequently in suppressing the hypoxic response. In this context, the accumulation of indoxyl sulfate (an uremic toxin) in the systemic circulation due to a reduced renal clearance, resulting in renal and cardiovascular dysfunction[56], was due to CITED2 stabilization and consequent HIF-1α inhibition[55].Also, in a type 2 diabetes animal model, Wanget al[57]have shown that, although insulin is able to downregulate CITED2 in endothelial cells, the vascular insulin resistance characteristic of this disease contributes to the upregulation of CITED2,which impairs HIF signaling and, consequently aborts angiogenesis[57].
In cancer, HIF-1 activates the transcription of genes that are involved in crucial aspects of cancer progression, including angiogenesis, cell survival, glucose metabolism, invasion and cell self-renewal[58,59]. In several types of cancer,intratumoral hypoxia and genetic alterations were reported to lead to HIF-1α overexpression, which is associated with increased patient mortality. Therefore, the inhibition of HIF-1 activity is regarded as a promising therapeutic approach[22,58].Importantly, the intricate HIF pathway also initiates anti-tumorigenic mechanisms that lead to cell cycle arrest or cell death[60,61], illustrating the need for a stringent control of the hypoxia response. Thus, HIF feedback regulators help to adjust and adapt HIF-activated responses to the fluctuating oxygen concentrations within tumors and to restrict the tumor-suppressing components of the HIF pathway. Therefore,given their role in cancer biology, HIF feedback regulators such as CITED2 may represent attractive targets for cancer therapy[62].
CITED2 AND HYPOXIA IN CANCER
The potential involvement of CITED2 in cancer was unveiled since it was demonstrated to be a transforming gene in Rat1 cells. Indeed, when overexpressed, Cited2 induced loss of cell contact inhibition, anchorage-independent growth, and tumor formation in nude mice[2]. Since this first publication describing CITED2, the dysregulation of its expression has been widely associated with aggressiveness and prognosis of several cancers including, among others, breast, colon, prostate and gastric cancers, as well as acute myeloid leukemia.
CITED2 in breast cancer
Breast cancer (BC) is the most frequent cancer affecting women worldwide, and the second leading cause of cancer-related deaths in women[63]. Studies using both animal models and human BC primary samples showed that CITED2 expression was elevated in primary tumors and metastasis, when compared to normal mammary epithelium[64,65]. However, the reports on CITED2 function and prognostic in BC are discrepant. Indeed, high CITED2 mRNA levels were associated with a clinical benefit in tamoxifen-treated BC and a prolonged metastasis-free survival in patients who had not received adjuvant systemic therapy[66]. More recent studies, showed that high CITED2 levels in primary tumors, when compared to normal mammary epithelium,were inversely correlated with patient survival[64,67]. In addition, high levels of CITED2 expression were shown to significantly increase the proliferation and migration of MCF-7 and SKBR-3 BC cell lines in culture[65]. By studying the possible mechanism involved in tumor growth and metastization, Jayaramanet al[68]have shown that the expression of IKKα and other NF-κB targets, with recognized roles in the metastatic process, were significantly decreased in both MDA-MB-231 and MDA-MB-468 cell lines followingCITED2knockdown. Moreover, the restoration of IKKα expression in CITED2-depleted cells, restored their invasive capacity[68]. In addition,CITED2silencing was also associated with reduced primary tumor growth, influencing the tumor vasculature by preventing TGF-β induction of VEGFAviaCITED2 recruitment to theVEGFApromoter[69]. Interestingly, the authors reported that HIF-1α was not involved in this process[69]. This is a surprising observation, since increasing VEGFA expression and tumor vasculature are prime roles usually attributed to HIF-1α and expected to be counteracted by CITED2. Therefore, this report supports the notion that negative regulation of HIF-1α by CITED2 may be cell-type dependent or somehow impaired in breast cancer cells. Alternatively, CITED2 may in this case act as co-activator of other transcription factors, such as SMAD2/3[18], which are mediators of TGF-β signaling pathway, or TFAP2A, showed to cooperate with CITED2 for normal vascularization of the myocardium during heart development[70].Other studies also reported a reduction in primary tumor growth due to the attenuation of tumor-associated macrophage (promoting tumor development and progression) recruitment in response to CITED2 depletion in cancer cells and the consequent downregulation of the macrophage chemoattractant CCL20[71]. This suggests that CITED2 promotes tumor-associated macrophage recruitment and infiltration in breast tumors. Metastases are the ultimate cause of death in BC patients and have a special tropism to develop in the bone. Although the molecular and cellular mechanisms behind BC cell homing and colonization of the bone are not fully understood, it was shown that intracardiac injection of CITED2-depleted NT2.5 mammary tumor cells in neu-N immunocompetent mice inhibited the establishment of bone metastasis and osteolysis, suggesting that CITED2 can promote osteotropism in BC[67]. CITED2 was also implicated in the acquisition of resistance to epirubicin and 5-fluorouracil therapies by inhibiting p53 accumulation[65], as well as resistance to tamoxifen, because it is a transcriptional co-activator of the estrogen receptor in breast cancer cells[64].
Altogether, these studies show that CITED2 is overexpressed in BC, contributing to prognosis, invasion and responsiveness to therapy. Moreover, CITED2 was shown to be induced by FOXO3a and to act as a transcriptional co-factor regulating HIF1-induced apoptosis in mouse embryonic fibroblasts and MCF-7 BC cells[72].Nevertheless, a role for CITED2 in mediating HIF effects in the context of BC was not clearly reported. Interestingly, its family member, CITED4, was found to be expressed in human BC cell lines and to be inversely associated with HIF-1α activity[73]. In fact,CITED4 was shown to be either expressed at low levels in the nucleus or trapped in the cytoplasm during breast tumor progression, implying in both circumstances that CITED4 had lost its ability to inhibit HIF-1α transcriptional activity, allowing the progression of the tumor size, grade and angiogenesis[73].
Cited2 and colorectal cancer
Colorectal cancer (CRC) is the second most deathly cancer worldwide[74]. CRC usually metastasizes to the liver, which is the cause of high mortality rates. In contrast to what was reported for BC, CITED2 depletion in CRC was associated with enhanced cell invasiveness[75]. Baiet al[75]have shown increased matrix metalloproteinase 13 (MMP-13) expression following CITED2 knockdown in RKO and SW480 CRC cell lines,suggesting that CITED2 may downregulate MMP-13 expression and limit invasiveness in CRC[75]. Notwithstanding, neutralizing MMP-13 activity with a monoclonal antibody, only slightly reduced the invasive capacity of SW480 cells expressing reduced levels of CITED2, suggesting that other changes in CITED2 knockdown cells also contributed to the altered invasiveness of these cells[75].
These data suggest that downregulation of CITED2 might have an important role in CRC progression but the mechanism mediating these effects is not fully known.Therefore, further studies should be undertaken to assess how CITED2 expression is modulated and whether low levels of CITED2 expression lead to increased HIF-1α transcriptional activity. Also using the SW480 cell line, Rogerset al[76]reported that CITED4 gene silencing modulated adherens/tight junction gene expression and reduced cell proliferation, without affecting apoptosis, colony formation, migration,invasion or adhesion[76].
Cited2 and prostate cancer
Prostate cancer (PC) is the most frequent cancer in men. More than half of the patients with PC present a translocation originating a fusion between the androgen-responsive gene TMPRSS2 and ETS (erythroblast transformation-specific)-family transcription factor genes such as ERG (ETS-related gene) and ETV1[77]. It has been speculated that CITED2 might be involved in PC, since its expression can be activated by ELK-1,another ETS family member[46], that is able to recruit androgen receptor to promote PC cell growth[78]. By testing this hypothesis, Shinet al[79]found that ERG was specifically upregulated as a consequence of the TMPRSS2–ERG gene fusion in PC cells.Interestingly, this fusion also upregulated CITED2, which was reported to promote post-translational modifications in nucleolin to enhance the metastatic potential of PC cells[79]. The metastatic facilitation occurs through AKT upregulation and a consequent increase of the epithelial–mesenchymal transition and invasion potential[79]. Therefore,CITED2-nucleolin-AKT signaling pathway should be considered as a potential target for therapies aiming to treat PC and prevent metastasis. A role for CITED2 in the regulation of HIF-1α in prostate cancer was not evaluated althoughin vitrostudies have previously shown that CITED2 is highly induced by hypoxia in DU145 prostate carcinoma cells[80].
Cited2 and gastric cancer
Gastric cancer (GC) is a silent and slow developing cancer that usually remains undetected until it reaches advanced stages[81]. The studies of Tanget al[82]using both GC cell lines and human primary samples showed that those cells can be categorized based on the CITED2 expression levels. In addition, CITED2 knockdown in cell lines with high CITED2 expression led to a decrease of their proliferation, mitochondrial membrane potential, colony formation, and an induced cell cycle arrest and apoptosis[82]. These results suggest that CITED2 can be considered as a good target for therapy in GC. Additionally, it was reported that GC cells with low expression of CITED2 are chemoresistant to anthracyclines. Interestingly, the pretreatment of GC cells with low expression of CITED2 with LBH589, an HDAC inhibitor, could reactivate the expression of CITED2 and sensitized them to chemotherapeutic drugs[83]. HIF-1α has been shown to be involved in various processes in GC[84], but the impact of CITED2 modulation on its activity remains to be established.
Cited2 and hepatocellular carcinoma
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related deaths worldwide[85]. CITED2 plays an important role in liver development, as indicated by knockouts ofCITED2in mice, which present fetal liver hypoplasia, due to increased cell apoptosis and disrupted cell-to-cell contact[86]. In HCC cells, CITED2 was identified as a direct activator of PPARγ, which possesses a tumor suppressive activity. CITED2 was significantly downregulated in primary HCCs when compared with their adjacent non-tumor tissues. In addition, CITED2 knockdown in the hepatocyte cell line LO2 and the HCC cell line Hep3B significantly increased cell viability and clonogenicity[87]. This was attributed to an increased cell cycle transition from G1 to S phase, concomitant with the downregulation of the cyclin-dependent kinase inhibitors p15INK4B, p21Wat1/Cip1, and p27Kip1and the upregulation of cyclin D1 expression[87]. In contrast, overexpression of CITED2 in HepG2 and BEL7404 HCC cell lines significantly suppressed cell growth by increasing p21Wat1/Cip1and p27Kip1[87].MicroRNAs have been reported to be promising biomarkers for HCC[88]. miR-1468, a novel cancer-related MicroRNAs which overexpression was associated with a poor prognosis in HCC patients, was reported to inhibit PPARγ/AKT signaling pathway through direct suppression of CITED2 and Up-frameshift protein 1, consequently promoting cell proliferation[89]. Jianget al[90]also found that miR-182-5p is upregulated in liver cell lines exposed to the human carcinogen trichloroethylen, which in turn inhibits CITED2 and enhances cell proliferation[90]. These data reinforce the antiproliferative effect of CITED2 in HCC. Interestingly, CITED2 was found to be degradedviathe ubiquitin-proteasome system and thus to be stabilized by proteasome inhibition[91]. Therefore, although HIF-1α can be upregulated by proteasome inhibition as shown in Hep3B cells and HEK293 embryonic kidney cells,its activity is reduced due to CITED2 interference with HIF-1α binding to p300 in hypoxic conditions[91]. This mechanism sheds light on how proteasome inhibitors may inhibit HIF-1α, which can be used as an anticancer therapy. Despite the knowledge of this important role for CITED2 in mediating the paradoxical responses of HIF-1α to proteasome inhibition, the regulation of the hypoxic response by CITED2 and its contribution to the pathogenesis of HCC remains elusive.
Cited2 and acute myeloid leukemia
Acute myeloid leukemia (AML) is a heterogeneous disease in which genetic and epigenetic factors contribute to the abnormal proliferation and differentiation of myeloblasts, generating a clonal population in the bone marrow[92]. An important regulator of hematopoiesis is the PU.1 transcription factor, which is expressed at low levels in hematopoietic stem and progenitor cells[93]and is crucial for hematopoietic stem and progenitor cells maintenance and differentiation of the myeloid lineage[94-98].In AML, PU.1 is usually dysregulated by mutations, translocations, and alterations in signal transduction, which promotes the accumulation of immature blasts[99,100]. PU.1 negatively regulates the expression of CITED2, by binding to multiple ETS-binding sites in the CITED2 promoter[101]. As shown by conditional knockout studies, Cited2 is essential for HSC maintenance. In contrast, the lack of Cited2 was not so evident in more committed cells[52]. Anderssonet al[102]have shown that AML cells have high levels of CITED2 expression[102]. In addition, Korthuiset al[101]have shown that CITED2 overexpression alone is enough to maintain the primitive CD34+CD38−HSC pool by decreasing apoptosis and enhancing quiescence, proving that CITED2 is crucial for AML maintenance[101]. More recently, Matteset al[103]have reported that loss of CITED2 impairs AML cell survivalviap53-mediated apoptosis by interfering with the AKT signal transduction pathway. Moreover, the same authors have shown that simultaneous upregulation of CITED2 and downregulation of PU.1 enhances the lifespan of HP hematopoietic stem and progenitor cells (HPSCs), which makes them more prone to full leukemic transformation[104]. These results indicate that CITED2 plays an important role in the pathogenesis of AML and it should be considered as a target for AML therapy. Interestingly, HIF-1α was reported to play also important roles in the self-renewal of AML leukemic stem cells and was indicated as a potential therapeutic target for eliminating these leukemic stem cells[105-107], but a possible role for CITED2 in this context was not explored.
Cited2 and lung cancer
Lung cancer is the leading cause of cancer-related death and approximately 80% of lung cancers are identified as non-small cell lung carcinoma (NSCLC)[108]. It is known that TGF-α and TGF-β are two key cytokines that regulate proliferation and quiescence, respectively, of lung epithelial cells during both normal and neoplastic lung development[109]. CITED2 plays also an important role in lung development, as demonstrated by the fact thatCITED2knockout mice display abnormal fetal lung development[13]. Chouet al[110]showed that in lung cancer, CITED2 works as a molecular switch for TGF-α proliferation and TGF-β quiescence stimuli in a MYCdependent pathway. CITED2 was shown to recruit p300 to induce MYC-p300-mediated transactivation of E2F3, leading to increased G1 to S cell cycle progression.Moreover, CITED2 was able to inhibit cellular quiescence by promoting MYCmediated suppression of p21CIP1[110]. Moreover, the same authors also observed that CITED2/MYC/E2F3/p21CIP1pathway was activated in patients with NSCLC with a poor prognosis[110].
Cited2 and cancer stem cells
The previous studies support the notion that CITED2 has context-dependent roles in cancer (Table 1). Although less extensively, CITED2 has also been shown to play a role in some other cancers, such as undifferentiated pleomorphic sarcoma,osteosarcoma, thyroid, and ovarian cancers[111-114]. CITED2 is also likely to play a role in an important aspect of tumor development related to cancer stem cells (CSCs). It is currently accepted that tumors originate from CSCs, which may derive from normal stem or progenitor cells that have lost the ability to self-regulate proliferation and quiescence[115,116]. Interestingly, an abnormal expression of core pluripotency transcription factors, such as OCT4, SOX2, NANOG, TBX3 and KLF4, has also been associated with CSCs, suggesting that the expression of pluripotent gene regulatory network factors may contribute to the conversion of normal cells into CSCs[117]. In murine embryonic stem cells, Cited2 controls the expression of Nanog, Tbx3, Klf4 and Oct4[118,119], and in adult stem cells it was associated with self-renewal, survival and quiescence[52,120,121]. Moreover, the CITED2-target geneBMI1, was shown to be involved in various CSC functions[122-124]. Thus, in particular circumstances, an anomalous increase of CITED2 expression may contribute to uncontrolled self-renewal and proliferation of stem cells and originate CSCs. Supporting this notion, a subset of patients with AML present an aberrantly elevated expression of CITED2 in CD34-positive leukemic cells compared to normal cells[101]. The imbalanced CITED2 expression due to a failure of PU.1 repression during normal myelopoiesis is likely to promote the initiation and maintenance of leukemia, and potentiate the establishment of a subset of multipotent leukemic stem cells[101]. On the other hand, patients with NSCLC expressing CITED2/MYC/E2F3/p21CIPhave a poor prognosis[110], but CITED2 was demonstrated to repress the expression of CSCs markers in NSCLC-stem cells and enhance their sensitivity to ionizing radiations in combination with butyrate treatment[125]. Therefore, the potential role played by CITED2 in the generation and maintenance of CSCs may vary with the nature of the tumor. Interestingly, increasing evidence indicates that HIFs regulate the sub-populations of CSCs in BC, CRC, and AML, for instance[126]. Therefore, studies to determine whether abnormal levels of CITED2 are important in CSC functions and weather CITED2-mediated inhibition of HIF signaling is on the basis of these functions should be pursued.
CONCLUSION
Altogether, the previous studies have shown that CITED2 is expressed ubiquitously and exhibits a very high affinity for the CH1 domain of the transcriptional coactivators CBP/p300, for which it competes with HIFs. CITED2 plays an important role in inhibiting HIF-1α in some diseases reviewed here, and may play a role in many others, since altered HIF activity was reported also in stroke, heart attack, and pulmonary hypertension, for example. In the particular case of cancer, CITED2 has been reported to have both oncogenic and tumor suppressive properties, depending on the cell/tumor type and treatment, like it was also shown for HIF. A role for CITED2 in the maintenance of CSCs was also unveiled in some cancers and seems to be also context-dependent. On the other hand, HIF signaling in CSCs is well established. Despite evidence linking both CITED2 and HIF functions independently to several aspects of cancer, little data linking the interplay between these two factorsin these processes is available. Therefore, comprehensive studies exploring the interactions between CITED2 and HIF-1α, which are important mediators in cancer,are sorely needed. A better understanding of this interplay may potentially lead to novel strategies for the development of innovative, targeted therapies.
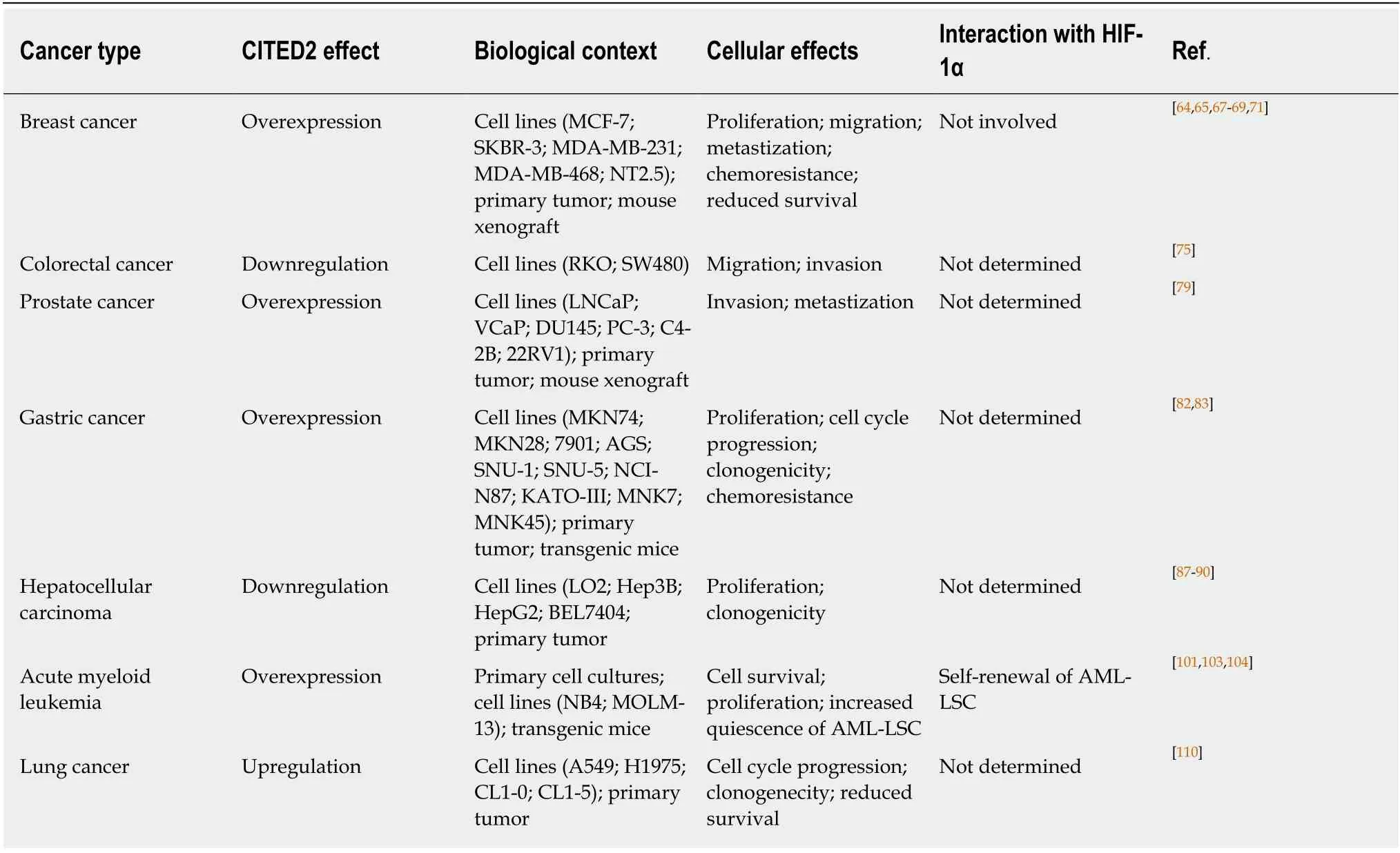
Table 1 Effects of CITED2 in different cancer
 World Journal of Clinical Oncology2020年5期
World Journal of Clinical Oncology2020年5期
- World Journal of Clinical Oncology的其它文章
- Formulation strategies in immunotherapeutic pharmaceutical products
- Impact of primary tumour location on colorectal liver metastases: A systematic review
- Human epidermal growth factor receptor 2 positive rates in invasive lobular breast carcinoma: The Singapore experience
- Immunotherapy – new perspective in lung cancer