
LGK974的合成工艺研究
2020-06-04 12:24:50骆荣双黄朋越刘春江赵庆平周志旭黄筑艳
合成化学 2020年5期
骆荣双, 黄朋越, 刘春江, 赵庆平, 周志旭, 黄筑艳*
(1. 贵州大学 a. 药学院; b. 贵州省合成药物工程实验室; c. 化学与化工学院,贵州 贵阳 550025)
Wnt信号在细胞增殖、分化和胚胎形态变化过程中起着主导作用,阻断Wnt信号传导是抗癌疗法中极具吸引力的方案之一[1-2]。LGK974是一种小分子Porcupine蛋白抑制剂,通过抑制Porcupine的活性控制Wnt蛋白的转运,阻断Wnt信号通路,可达到抗癌效果[3-6]。
文献合成LGK974的方法为[7-8]:以2-氯-3-甲基-5-溴吡啶、2-叔丁氧基-2-羰基乙基锌氯、2-甲基-4-(三丁基锡)吡啶、2-氨基-5-吡啶硼酸酯、2-碘吡嗪为原料,经5步反应合成目标化合物,总收率14.6%。该合成路线的缺点较多,如使用的原料2-碘吡嗪、2-氨基-5-吡啶硼酸酯价格昂贵;中间产物6-氯-5-甲基-3-吡啶乙酸叔丁酯需使用硅胶柱层析纯化;有机锡试剂具有较强的神经毒性,难以去除,对药效产生不良影响。
为了优化LGK974的合成路线,本文以2-氨基-5-溴吡啶、2-氯-3-甲基吡啶、2-氨基吡嗪、2-甲基-6-氯吡啶为起始原料,经取代、Suzuki、酰胺化等10步反应合成了LGK974(Scheme 1),总收率11.5%,其结构经1H NMR、13C NMR和MS(ESI-TOF)确证。
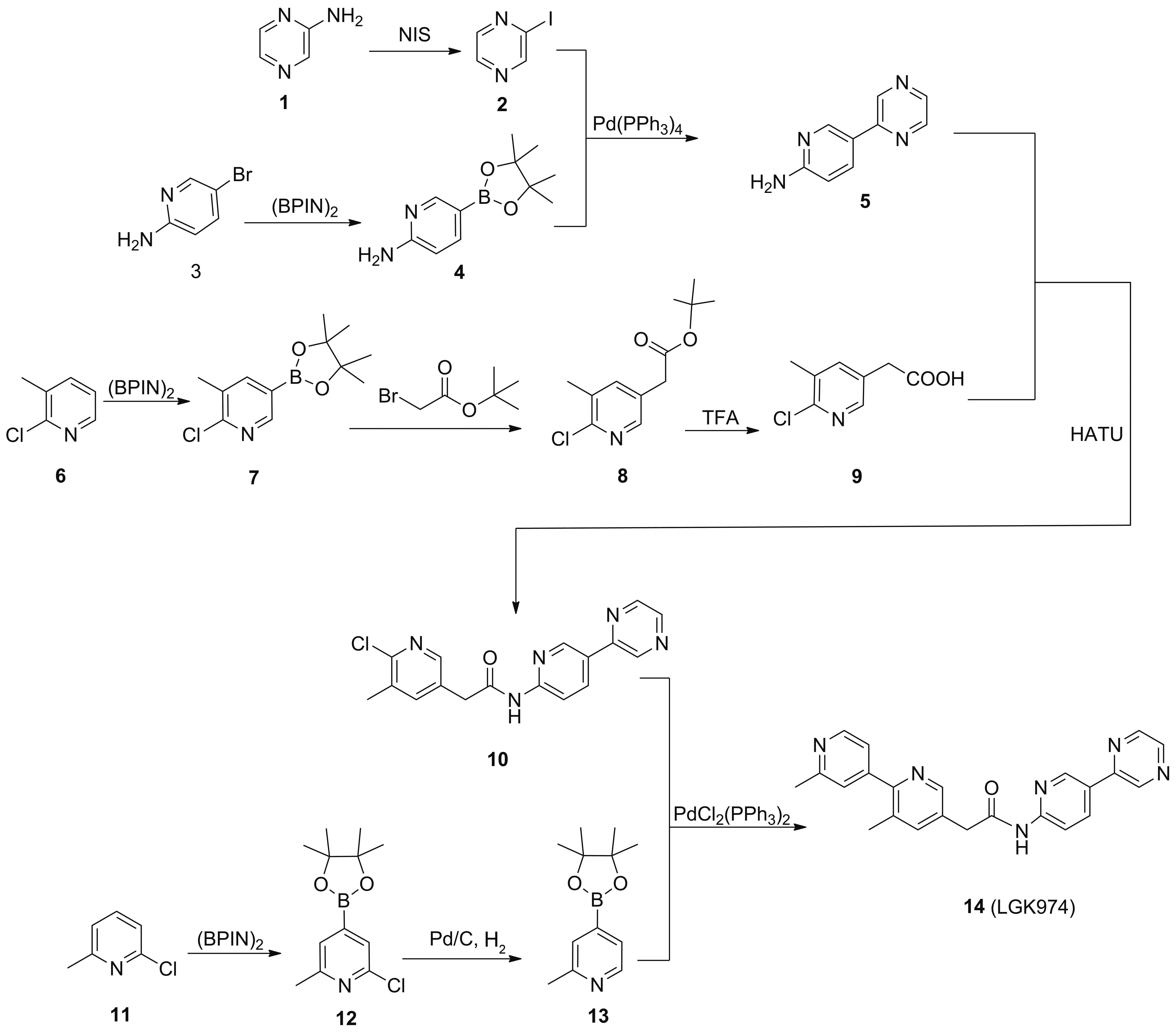
Scheme 1
1 实验部分
1.1 仪器与试剂
T-4型熔点仪;ZF-7型三用紫外分析仪;Bruker Advance DMX 400 MHz型核磁共振仪(TMS为内标);Agilent 1100 LC/MS型质谱仪。
所用试剂均为化学纯或分析纯。
1.2 合成
(1) 2-碘吡嗪(2)的合成
在1000 mL单口烧瓶中加入2-氨基吡嗪(1)100.2 g(1051.4 mmol)和N,N-二甲基甲酰胺400 mL,于0 ℃缓慢加入亚硝酸钠109.3 g(1577.1 mmol)和N-碘代丁二酰亚胺(NIS)355.1 g(1577.1 mmol),升温至室温,搅拌下反应4 h。缓慢倒入适量冰水中,用乙酸乙酯(3×200 mL)萃取,合并有机相,依次用饱和硫代硫酸钠溶液洗涤,无水硫酸钠干燥,浓缩得黄色油状液体2191.1 g,收率88.1%;1H NMR(CDCl3, 400 MHz)δ: 8.40(dd,J=1.8 Hz, 2.4 Hz, 1H), 8.52(d,J=2.4 Hz, 1H), 8.64(d,J=1.5 Hz, 1H)。
(2) 2-氨基-5-吡啶硼酸酯(4)的合成
在500 mL三口瓶中加入2-氨基-5-溴吡啶(3)54.5 g(314.5 mmol)、联硼酸频哪醇酯(BPIN)279.0 g(330.0 mmol)、醋酸钾92.5 g(943.5 mmol), [1,1′-双(二苯基膦)二茂铁]二氯化钯11.5 g(15.8 mmol)和1,4-二氧六环300 mL,氮气保护下,升温至100 ℃,搅拌下反应8 h。抽滤,滤饼用甲醇洗涤,合并滤液和洗液,旋蒸除溶,残余物用甲醇浸泡后抽滤,滤液加入活性碳,搅拌5 h,抽滤,滤液蒸除溶剂后经正己烷打浆得白色固体464.2 g,收率92.6%, m.p.98.3~101.6 ℃;1H NMR(400 MHz, CDCl3)δ: 1.35(s, 12H), 4.47(s, 2H), 6.43(d,J=8.0 Hz, 1H), 7.77(d,J=10.0 Hz, 1H), 8.44(s, 1H)。
(3) 5-(2-吡嗪基)-2-吡啶胺(5)的合成
在500 mL三口瓶中加入2-碘吡嗪50.0 g(242.5 mmol)、 2-氨基-5-吡啶硼酸酯25.6 g(291.5 mmol)、碳酸钾100.0 g(727.5 mmol)、四(三苯基膦)钯[Pd(PPh3)4]8.0 g(7.3 mmol)和混合溶剂[V(1,4-二氧六环)/V(水)=10/1]300 mL,氮气保护下,升温至 80 ℃,搅拌下反应8 h。减压浓缩,残余物加入丙酮50 mL,抽滤,滤液浓缩,粗品加入乙酸乙酯100 mL,抽滤得黄白色固体532.7 g,收率78.3%, m.p.125.2~127.6 ℃;1H NMR(CDCl3, 400 MHz)δ: 9.12(d,J=1.6 Hz, 1H), 8.72(m, 1H), 8.59(m, 1H), 8.45(d,J=2.8 Hz, 1H), 8.11(dd,J=8.8 Hz, 2.4 Hz, 1H), 6.54(d,J=8.8 Hz, 1H), 6.44(s, 2H)。
(4) 6-氯-5-甲基-3-吡啶乙酸(9)的合成
在500 mL三口瓶中加入2-氯-3-甲基吡啶(6)60.1 g(469.8 mmol)、联硼酸频哪醇酯110.1 g(469.8 mmol)、甲氧基(环辛二烯)合铱二聚体0.3 g(0.5 mmol)、 4,4′-二叔丁基-2,2′-二吡啶0.6 g(2.3 mmol)和正己烷300 mL,氮气保护下,升温至60 ℃,搅拌下反应3 h。减压浓缩,残余物加入适量水中,抽滤,滤饼加入50 mL正己烷,搅拌0.5h得黄白色固体7117.0 g,收率98.3%, m.p.102.6~105.8 ℃;1H NMR(CDCl3, 400 MHz)δ: 8.56(d,J=1.8 Hz, 1H), 7.92(m, 1H), 2.39(s, 3H), 1.28(s, 12H)。
在500 mL三口瓶中加入2-氯-3-甲基-4-吡啶硼酸酯60.7 g(470.4 mmol)、磷酸钾150.6 g(710.1 mmol)、三(邻甲苯基)膦6.3 g(23.7 mmol)、溴乙酸叔丁酯64.1 g(284.1 mmol)、三(二亚苄基丙酮)二钯6.6 g(7.1 mmol)和混合溶剂[V(四氢呋喃)/V(水)=10/1]300 mL,氮气保护,搅拌下反应12 h。过滤,滤饼用少量四氢呋喃洗涤,合并滤液和洗液,减压浓缩,残余物用混合溶剂[V(正己烷)/V(乙酸乙酯)=10/1]浸泡,抽滤,滤液减压浓缩,残余物加入二氯甲烷100 mL和三氟乙酸135.2 g(1183.5 mmol),反应2 h。加入冰水,用二氯甲烷(3×200 mL)萃取,合并有机相,依次用饱和碳酸氢钠溶液200 mL洗涤,无水硫酸镁干燥,浓缩得黄色固体920.1 g,收率45.2%, m.p.120.4~123.1 ℃;1H NMR(CDCl3, 400 MHz )δ: 8.19(m, 1H), 7.56(m, 1H), 3.65(s, 2H), 2.40(s, 3H)。
(5) 6-氯-5-甲基-N-[5-(2-吡嗪基)- 2-吡啶基]-3-吡啶乙酰胺(10)的合成
在500 mL三口瓶中加入5-(2-吡嗪基)-2-吡啶胺29.8 g(174.0 mmol)、 6-氯-5-甲基-3-吡啶乙酸32.4 g(174.0 mmol)、 2-(7-偶氮苯并三氮唑)-N,N,N′,N′-四甲基脲六氟磷酸酯(HATU)66.2 g(174.0 mmol)、三乙胺19.2 g(191.4 mmol)和N,N-二甲基甲酰胺200 mL,搅拌下反应10 h。抽滤,滤液倒入适量水中,用乙酸乙酯(3×100 mL)萃取,合并有机相,依次用饱和食盐水洗涤,无水硫酸钠干燥,减压浓缩,残余物加入乙醚50 mL,抽滤得白色固体1044.4 g,收率75.5%, m.p.223.2~226.5 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 10.39(s 1H), 9.31(d,J=1.6 Hz, 1H), 9.11(d,J=1.6 Hz, 1H), 8.72(m, 1H), 8.65(m, 1H), 8.52(dd,J=8.6 Hz, 2.4 Hz, 1H), 8.20(m, 2H), 7.73(d,J=1.6 Hz, 1H), 3.91(s, 2H), 2.34(s, 3H)。
(6) 2-甲基-4-吡啶硼酸酯(13)的合成
在500 mL三口瓶中加入2-甲基-6-氯吡啶(11)60.0 g(470.4 mmol)、联硼酸频哪醇酯110.1 g(156.8 mmol)、甲氧基(环辛二烯)合铱二聚体0.3 g(0.5 mmol)、 4,4′-二叔丁基-2,2′-二吡啶0.6 g(2.3 mmol)和正己烷300 mL,氮气保护下,升温至60 ℃,搅拌下反应2.5 h。减压浓缩,残余物倒入适量水中,抽滤,滤饼用少量水洗涤,过滤,滤饼加入正己烷50 mL,抽滤得白色固体12107.4 g,收率90.0%, m.p.105.2~107.8 ℃;1H NMR(CDCl3, 400 MHz)δ: 8.56(d,J=1.8 Hz, 1H), 7.92(m, 1H), 2.35(s, 3H), 1.34(s, 12H)。
在500 mL三口瓶中加入2-甲基-6-氯-吡啶硼酸酯29.7 g(118.4 mmol)、甲酸铵15.1 g(236.6 mmol)、钯炭3.0 g(wt=5%)和甲苯200 mL,氮气保护,搅拌下反应6 h。过滤,滤液减压浓缩,残余物加入丙酮100 mL,搅拌0.5 h;抽滤得白色固体1321.8 g,收率84.9%, m.p.59.6~62.5 ℃;1H NMR(CDCl3, 400 MHz)δ: 8.51(dd,J=4.8 Hz, 1.0 Hz, 1H), 7.51(s, 1H), 7.42(d,J=4.8 Hz, 1H), 2.55(s, 3H), 1.34(s, 12H)。
(7) LGK974(14)的合成
在500 mL三口瓶中加入6-氯-5-甲基-N-[5-(2-吡嗪基)-2-吡啶基]-3- 吡啶乙酰胺29.4 g(88.2 mmol)、磷酸钾56.4 g(264.6 mmol)、 2-甲基-4-吡啶硼酸酯22.8 g(105.6 mmol)、双(三苯基膦)二氯化钯[PdCl2(PPh3)2]1.8 g(14.2 mmol)和混合溶剂[V(1,4-二氧六环)/V(水)/V(乙醇)=20/1/0.5]300 mL,氮气保护,搅拌下回流反应12 h。过滤,滤饼用少量甲醇洗涤,合并滤液和洗液,减压浓缩,残余物加入少量水,抽滤,滤饼加入乙醚50 mL,搅拌1 h;抽滤得黄白色固体1424.1 g,收率70.3%, m.p.263.8~266.9 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 11.08(s 1H), 9.30(d,J=1.6 Hz, 1H), 9.11(d,J=1.6 Hz, 1H), 8.63(m, 1H), 8.59(m, 1H), 8.47(dd,J=8.6 Hz, 2.4 Hz, 1H), 8.13(m, 3H), 7.89(m, 1H), 7.77(m, 1H), 7.70(m, 1H), 3.83(s, 2H), 2.34(s, 3H), 2.32(s, 3H);13C NMR(DMSO, 100 MHz)δ: 168.2, 159.2, 156.8, 156.6, 150.7, 148.4, 148.34, 147.1, 144.3, 143.1, 142.2, 141.4, 133.6, 132.3, 131.3, 130.8, 130.6, 122.1, 119.7, 112.7, 41.8, 23.8, 18.6; HR-MS(ESI-TOF)m/z: Calcd for C23H20N6O{[M+H]+}397.1771, found 397.1774。
2 结果与讨论
2.1 化合物10的合成[9-10]
考察了不同酰胺化方法对反应的影响,结果见表1。由表1可知,选择HATU/TEA体系时,收率最高为75%。

表1 酰胺化方法对化合物10收率的影响
2.2 化合物13的合成
合成化合物13时,以2-甲基-6-氯吡啶为起始原料,通过催化剂甲氧基(环辛二烯)合铱二聚体催化在吡啶环的4-位成功引入硼酸酯,在氢气、碱以及催化剂同时存在的条件下进行还原反应得到化合物13。对催化剂用量和碱对13收率的影响进行了优化。
(1) 催化剂用量
文献[11]方法表明,反应时需加入催化剂进行催化还原,催化剂用量对反应有较大的影响。对催化剂用量进行优化后发现,钯碳(wt=5%)催化剂与原料质量比为0.15/1时,收率最高为89.1%。
(2) 碱
文献[11]方法使用的碱包括碳酸氢钠、叔丁醇钾、碳酸钾、甲酸铵。在实验过程中发现,没有碱参与反应时,反应不能成功引发;随着碱性的增强,收率降低。采用甲酸铵为碱,收率最高为86.4%(表2)。

表2 碱对化合物13收率的影响
2.3 化合物14的合成
Suzuki偶联反应为构筑芳香环间C—C键的有效方法[12-15]。采用Suzuki偶联反应合成了化合物14,并研究了碱对收率的影响,结果见表3。由表3可知,以磷酸钾为碱时,收率最高为67.9%。

表3 碱对化合物14收率的影响
以2-氨基-5-溴吡啶和2-氨基吡嗪、2-甲基-6-氯吡啶等为起始原料,采用汇聚式合成法,经取代、Suzuki、酰胺化等10步反应合成了Porcupine抑制剂LGK974,总收率11.5%。该路线具有成本低、安全环保、副产物少及后处理简单等优点。
猜你喜欢
——非均布滤饼的局部比阻与平均比阻的测定与计算方法
化工装备技术(2020年4期)2020-09-09 07:34:10
流体机械(2020年5期)2020-06-24 05:39:08
设备管理与维修(2019年9期)2019-09-12 07:44:02
石油钻探技术(2018年5期)2018-10-13 07:25:18
中国矿业(2017年2期)2017-02-28 02:12:56
大连工业大学学报(2015年4期)2015-12-11 04:06:50
中国塑料(2015年7期)2015-10-14 01:02:48
中国酿造(2015年4期)2015-01-26 22:50:40
应用化工(2014年12期)2014-08-16 13:10:46
火炸药学报(2014年1期)2014-03-20 13:17:22
