
新型吲哚苯醌衍生物的合成及抗肿瘤活性
2020-06-04 12:24阮铱瞳
合成化学 2020年5期
阮铱瞳, 何 菱
(四川大学 华西药学院,四川 成都 610041)
醌类衍生物表现出良好抗肿瘤活性,尤其蒽醌类化合物是天然醌类结构中数量最多、应用最广、最受关注的化合物。其药理作用较为广泛,大多数蒽醌类化合物均具有抗菌抗炎、抗病毒、抗肿瘤作用,另外还可用于治疗慢性肾炎,抗衰老,抗诱变,降血脂等[1-4],如临床一线抗肿瘤药物米托蒽醌,阿霉素等(Chart 1)[5-8]。同时,具有“2-苯基萘型”结构单元的喜树碱和棕霉素等(Chart 1)也在临床应用中显示良好的抗肿瘤活性[9]。
基于吲哚醌类衍生物的多样生物活性[10-15],结合药物设计学中的拼合原理,我们认为组合后的新结构可能会具有抗肿瘤活性或新的生物活性。因此,本课题组在前期工作中,以“一勺烩”方式合成系列双取代相同吲哚苯醌衍生物和两步式合成双取代不同吲哚苯醌衍生物并进行了抗肿瘤活性研究[17]。并提出了改进文献[16]方法的建议:(1)不能一锅煮同时合成双吲哚取代苯醌衍生物;(2)添加相转移催化剂缩短反应时间;(3)虽然不能区域选择性获得2,5-双取代吲哚苯醌衍生物,但可通过柱层析快速分离,避免全合成路线过长和使用金属催化剂。由于抗肿瘤活性不理想,合成方法和底物拓展还有待进一步研究。
本文拟通过改变取代基改变其脂水分配系数,同时增加可供氢键形成的结合位点,希望改善抗肿瘤活性;并拟优化合成方法,进一步考察该合成方法的普适性,为生物活性筛选提供保障。因此,以2-取代吲哚衍生物(1a~1c)为底物,以无水乙腈为反应溶剂,在相转移催化剂苄基三乙基氯化铵(TEBA)和无水碳酸钾存在下,与四溴苯醌在室温及N2环境下反应,合成了9个未见文献报道的2-取代吲哚-1,4-醌衍生物(2a~2c,3a~3c,4a~4c, Scheme 1),其结构经1H NMR,13C NMR和HR-MS(ESI)表征。并进行了初步的抗肿瘤活性研究,发现化合物2b和3c对人乳腺癌细胞系MDA-MB-231具有良好的抑制活性,其IC50分别分别低于0.7 μmol·L-1和0.1 μmol·L-1,较课题组前期获得衍生物的抗肿瘤活性有所改善,为进一步构建抗肿瘤活性吲哚苯醌衍生物库和获得候选化合物建立基础。
1 实验部分
1.1 仪器与试剂
YRT-3型熔点仪;Varian Mercury 400 MHz/600 MHz型核磁共振仪(DMSO-d6, Methanol-d4或CD3Cl为溶剂,TMS 为内标);Bruker Daltonics Data analysis 3.2 Mass Spectrometer型质谱仪。
2-取代基吲哚(1a~1c)按文献[18-19]方法合成;乙腈,成都恒信化学试剂有限公司;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1)2a~2c的合成通法
称取化合物1a~1c0.33 mmol,四溴苯醌 65 mg(0.25 mmol),苄基三乙基氯化铵(TEBA)75mg(0.33 mmol)和无水K2CO345mg(0.33 mmol)置于密闭反应装置中,置换氮气,加入无水乙腈5 mL,室温搅拌2~6 h(TLC检测),待原料四氯苯醌反应完全,蒸除溶剂,再将残余物溶于乙酸乙酯,用饱和食盐水20 mL洗涤,乙酸乙酯(3×20 mL)萃取,合并有机层,经无水硫酸钠干燥,蒸除溶剂,残余物经硅胶柱层析[洗脱剂:V(石油醚)/V(乙酸乙酯)=10/1]纯化得2a~2c。
2,3,5-三溴-6-(2-甲基-5-三异丙基硅氧基)吲哚-1,4-对苯醌(2a): 蓝色固体131 mg,收率82%, m.p.132~134 ℃;1H NMR(400 MHz, CD3Cl)δ: 8.24(s, 1H), 7.14(d,J=8.6 Hz, 1H), 6.78(dd,J=8.6 Hz, 1.9 Hz, 1H), 6.69~6.56(m, 1H), 2.29(s, 3H), 1.26(t,J=7.4 Hz, 3H), 1.10(s, 18H);13C NMR(100 MHz, CD3Cl)δ: 174.5, 171.7, 150.5, 143.1, 138.3, 137.6, 133.1, 130.4, 127.1, 116.0, 111.2, 109.8, 107.1, 18.0, 14.2, 12.7; HR-MS(ESI)m/z: Calcd for C24H29NO3Br3Si{[M+H]+}643.9461, found 643.9458。
2,3,5-三溴-6-[2-(三异丙基硅基甲基醚)吲哚]-1,4-对苯醌(2b): 蓝色固体136 mg,收率85%, m.p.127~130 ℃;1H NMR(400 MHz, CD3Cl)δ: 8.89(s, 1H), 7.46(d,J=8.1 Hz, 1H), 7.25(t,J=8.3 Hz, 2H), 7.20~7.15(m, 1H), 4.89(d,J=22.8 Hz, 2H), 1.18(dq,J=12.3 Hz, 6.9 Hz, 5.8 Hz, 3H), 1.09(d,J=5.2 Hz, 18H);13C NMR(100 MHz, CD3Cl)δ: 170.5, 167.5, 138.6, 135.9, 134.2, 130.6, 122.1, 118.4, 116.7, 116.4, 107.5, 100.5, 55.3, 49.4, 13.9, 7.9; HR-MS(ESI)m/z: Calcd for C24H28NO3SiBr3Na{[M+Na]+}665.9281, found 665.9275。

Chart 1
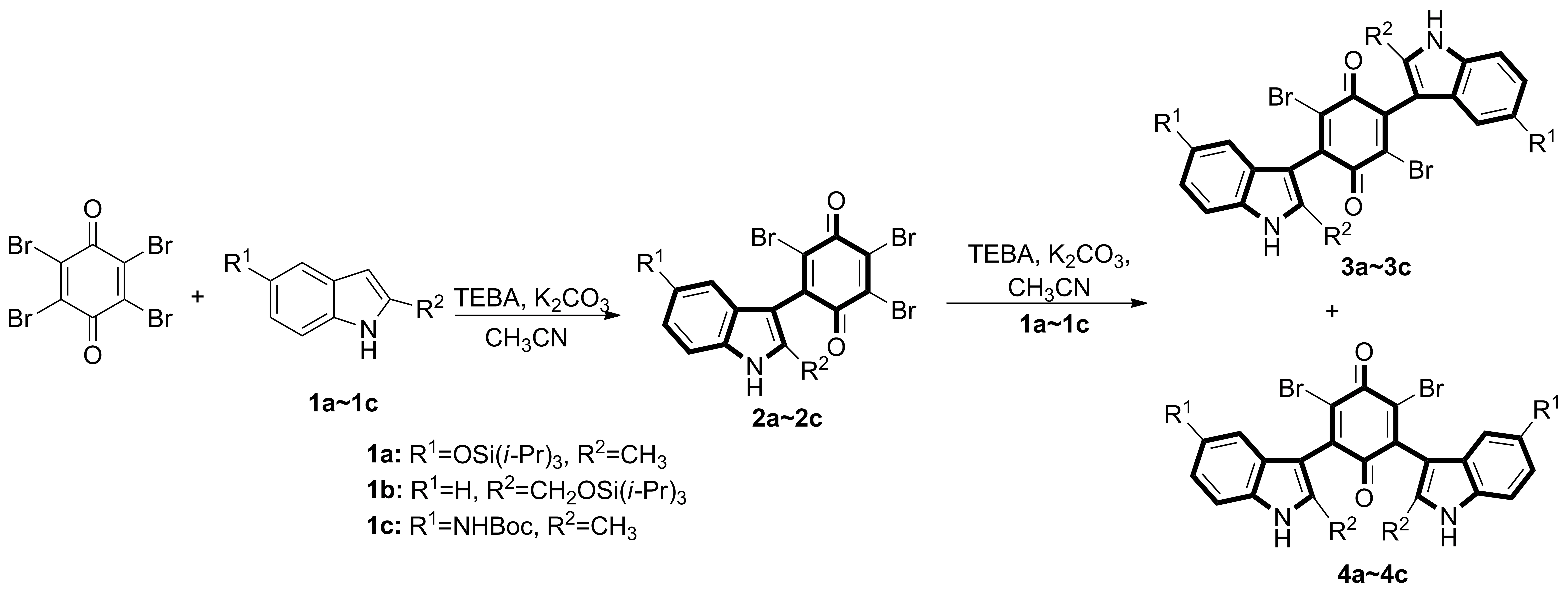
Scheme 1
2,3,5-三溴-6-[2-甲基-5-(N-叔丁氧羰基)吲哚]-1,4-对苯醌(2c):蓝色固体126 mg,收率86%, m.p.135~136 ℃;1H NMR(400 MHz, CD3Cl)δ: 8.74(s, 1H), 7.07(s, 2H), 6.60(s, 1H), 2.24(s, 3H), 1.53(s, 9H);13C NMR(100 MHz, CD3Cl)δ: 175.1, 171.8, 153.6, 141.3, 140.6, 139.5, 138.7, 138.6, 132.2, 131.6, 127.1, 111.3, 104.7, 60.5, 28.5, 13.9; HR-MS(ESI)m/z: Calcd for C20H17N2O3Br3K{[M+K]+}608.8631, found 608.8629。
(2)3a~3c和4a~4c的合成通法
称取2a~2c0.20 mmol,1a~1c0.24 mmol,苄基三乙基氯化铵55 mg(0.24 mmol)和无水K2CO332 mg(0.24 mmol)置于密闭反应装置中,置换氮气,加入无水乙腈 5 mL,搅拌下反应4~24 h(TLC监测)。蒸除溶剂,残余物依次用乙酸乙酯溶解,饱和食盐水(20 mL)洗涤,乙酸乙酯(3×20 mL)萃取,合并有机层,用无水硫酸钠干燥,蒸除溶剂,残余物经硅胶柱层析[洗脱剂:V(石油醚)/V(二氯甲烷)=1/1]纯化得3a~3c和4a~4c。
2,5-二溴-3,6-(2-甲基-5-三异丙基硅氧基)吲哚-1,4-对苯醌(3a): 蓝色固体52 mg,收率30%, m.p.140~142 ℃;1H NMR(400 MHz, CD3Cl)δ: 8.20(d,J=10.6 Hz, 2H), 7.16(d,J=8.6 Hz, 2H), 6.81~6.72(m, 4H), 2.38(d,J=6.2 Hz, 6H), 1.29(t,J=3.8 Hz, 6H), 1.11(d,J=7.3 Hz, 36H);13C NMR(100 MHz, CD3Cl)δ: 177.0, 177.0, 150.4, 150.4, 142.9, 142.7, 137.8, 137.8, 137.17, 137.15, 135.3, 135.2, 130.5, 130.4, 129.8, 127.5, 127.4, 127.1, 126.6, 125.4, 124.5, 123.9, 121.5, 121.1, 119.1, 115.8, 115.7, 111.0, 111.0, 110.0, 109.9, 107.4, 107.4, 105.0, 105.0, 18.0, 14.3, 14.1, 12.7; HR-MS(ESI)m/z: Calcd for C42H56N2O4Br2Si2Na{[M+Na]+}889.2038, found 889.2041。
2,6-二溴-3,5-(2-甲基-5-三异丙基硅氧基)吲哚-1,4-对苯醌(4a): 蓝色固体59 mg,收率34%, m.p.138~139 ℃;1H NMR(400 MHz, CD3Cl)δ: 8.13(s, 2H), 7.15~7.10(m, 2H), 6.78~6.68(m, 4H), 2.29(d,J=15.1 Hz, 6H), 1.20(dd,J=14.9 Hz, 7.2 Hz, 6H), 1.07(dd,J=21.6 Hz, 7.1 Hz, 36H);13C NMR(100 MHz, CD3Cl)δ: 180.2, 174.1, 174.0, 150.4, 150.3, 143.5, 139.8, 139.3, 137.9, 137.4, 137.2, 136.7, 134.0, 133.7, 133.6, 130.5,130.4, 127.8, 127.7, 127.5, 127.3, 124.5, 124.0, 119.1, 115.7, 115.6, 111.0, 110.9, 109.9, 107.5, 107.4, 105.4, 105.2, 18.0, 14.0, 12.7; HR-MS(ESI)m/z: Calcd for C42H56N2O4Si2Br2Na{[M+Na]+}889.2038, found 889.2041。
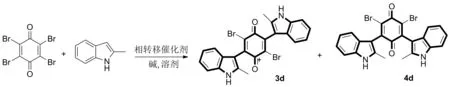
Scheme 2
2,5-二溴-3,6-二[2-(三异丙基硅基甲基醚)吲哚]-1,4-对苯醌(3b): 蓝色固体36 mg,收率21%, m.p.141~143 ℃;1H NMR(400 MHz, CD3Cl)δ: 8.33(s, 1H), 8.03(s, 1H), 7.54(s, 1H), 7.42(d,J=8.1 Hz, 1H), 7.22~7.14(m, 3H), 7.10~7.04(m, 3H), 4.93(s, 2H), 4.26(s, 2H), 1.21~1.13(m, 6H), 1.09(d,J=6.6 Hz, 36H);13C NMR(100 MHz, CD3Cl)δ: 147.7, 147.1, 138.6, 138.0, 136.0, 135.2, 134.9, 130.0, 128.9, 128.5, 124.5, 124.0, 122.0, 120.9, 119.8, 119.5, 119.1, 118.6, 111.0, 110.5, 107.3, 99.8, 57.6, 18.0, 11.9; HR-MS(ESI)m/z: Calcd for C42H56N2O4Si2Br2Na{[M+Na]+}889.2038, found 889.2039。
2,6-二溴-3,5-二[2-(三异丙基硅基甲基醚)吲哚]-1,4-对苯醌(4b): 蓝色固体40 mg,收率23%, m.p.140~141 ℃;1H NMR(400 MHz, CD3Cl)δ: 8.85(d,J=10.0 Hz, 2H), 7.45(dd,J=8.0 Hz, 4.5 Hz, 2H), 7.26~7.17(m, 6H), 4.32(s, 2H), 4.23(s, 2H), 1.20~1.16(m, 6H), 1.11~1.08(m, 36H);13C NMR(100 MHz, CD3Cl)δ: 177.7, 177.4, 174.7, 174.0, 157.2, 156.7, 142.2, 140.7, 140.0, 140.0, 139.1, 134.7, 132.0, 129.9, 126.3, 124.5, 124.0, 122.4, 122.3, 120.6, 120.2, 111.5, 59.4, 18.0, 11.9; HR-MS(ESI)m/z: Calcd for C42H56N2O4Si2Br2Na{[M+Na]+}889.2038, found 889.2039。
2,5-二溴-3,6-二[2-甲基-5-(N-叔丁氧羰基)吲哚]-1,4-对苯醌(3c): 蓝色固体 33 mg,收率22%, m.p.136~138 ℃;1H NMR(400 MHz, Methanol-d4)δ: 7.35(s, 1H), 7.20(d,J=8.4 Hz, 2H), 7.12(s, 1H), 7.01(d,J=8.5 Hz, 1H), 2.31(d,J=8.9 Hz, 6H), 1.48(d,J=11.0 Hz, 18H);13C NMR(100 MHz, DMSO-d6)δ: 177.2, 177.2, 153.6, 153.6, 143.0, 137.8, 137.7, 135.5, 132.4, 132.3, 132.1, 132.0, 127.1, 111.1, 107.2, 78.7, 28.7, 14.4, 13.9; HR-MS(ESI)m/z: Calcd for C34H34N4O6Br2Na{[M+Na]+}775.0737, found 775.0741。
2,6-二溴-3,5-二[2-甲基-5-(N-叔丁氧羰基)吲哚]-1,4-对苯醌(4c): 蓝色固体38 mg,收率25%, m.p.136~138 ℃;1H NMR(400 MHz, methanol-d4)δ: 7.35(s, 1H), 7.20(d,J=8.4 Hz, 3H), 7.12(s, 1H), 7.01(d,J=8.6 Hz, 1H), 2.31(d,J=8.1 Hz, 6H), 1.48(d,J=11.0 Hz, 18H);13C NMR(100 MHz, DMSO-d6)δ: 180.8, 180.3, 170.8, 153.4, 143.7, 137.8, 133.8, 133.6, 132.4, 132.4, 132.0, 131.9, 129.1, 127.3, 127.0, 111.1, 107.4, 107.1, 78.7, 28.7, 14.5; HR-MS(ESI)m/z: Calcd for C34H34N4O6Br2Na{[M+Na]+}775.0737, found 775.0745。
2 结果与讨论
2.1 合成
以2-甲基吲哚衍生物为底物在碱性条件以及相转移催化下与四溴苯醌缩合[17],为优化反应条件,提高双取代收率,以3d和4d的合成反应为模型反应(Scheme 2),对催化剂、溶剂、碱、反应温度进行了筛选。
(1) 催化剂
表1为催化剂的优化。由表1可见,相转移催化剂对反应影响的都能得到预期产物,但三乙基苄基氯化铵反应效果最好。无相转移催化剂双吲哚取代产物仅痕迹量,可获得部分单取代产物。于是作者选用三乙基苄基氯化铵作为催化剂进行进一步探索。
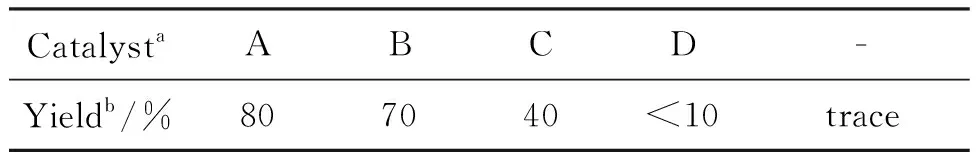
表1 催化剂的优化
aA~D:三乙基苄基氯化铵,四丁基氯化铵,18-冠-6,聚乙二醇400;b3+4,下同。
(2) 溶剂
表2为溶剂的优化。由表2可见,在甲苯、四氢呋喃溶液中只有微量产物,在DMF中反应较杂乱,在CH3CN 溶剂中反应效果最好。
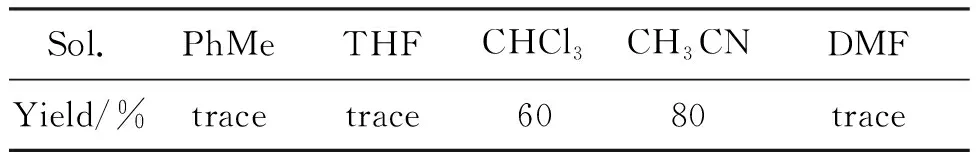
表2 溶剂的优化
(3) 碱
表3为碱对反应的影响。由表3可见,反应体系中不加碱,反应难以发生。在反应体系加入碱,对反应有明显的促进作用。加入碳酸钾和碳酸铯的收率相当,碳酸钾较廉价,最终选择碳酸钾为碱进行底物拓展反应。
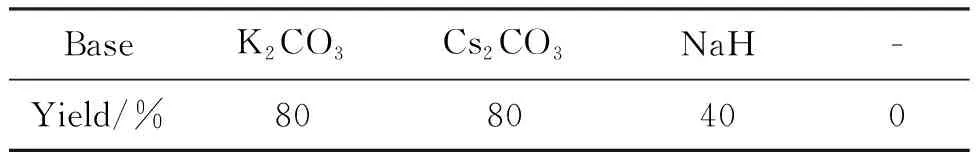
表3 碱对反应的影响
(4) 反应温度
表4为反应温度对收率的影响。由表4可见,随着反应温度的升高,收率逐渐降低。因此采用室温为反应温度。

表4 最佳反应温度
(5) 底物
作者对催化剂、溶剂、碱、反应温度进行筛选后,对底物的适应性进行了研究。对反应底物而言,仅当2-位有取代基(如苯基、烷基等衍生物)的吲哚衍生物才能获得双吲哚取代苯醌衍生物(投料比和反应时间可控制单取代和双取代),否则反应杂乱,收率低,难于分离,这也是该合成方法的不足之处。2-苯基(2-p-甲苯基)取代吲哚衍生物的反应性优于2-烷基取代吲哚衍生物。
2.2 体外细胞毒性
以苯达莫司汀(Bendamustiune,双功能烷化剂)和伏立诺他(SAHA, HDAC抑制剂)作为阳性药物对照,测试了目标化合物对人肺癌细胞系(A549)、人乳腺癌细胞(MDA-BM-231)和宫颈癌细胞系(Hela)的体外细胞毒性,结果见表5。由表5可知,所有化合物对A549, MDA-BM-231和Hela均有抑制活性。其中,化合物2b和3c对A549的IC50低于3 μmol·L-1,对MDA-BM-231的IC50低于0.7 μmol·L-1和0.1 μmol·L-1,对Hela的IC50低于2 μmol·L-1和0.4 μmol·L-1。

表5 化合物的体外抗肿瘤活性
以吲哚衍生物为底物,在相转移催化剂存在下,以碳酸钾为碱,在室温下与四溴苯醌反应,合成了9个新型的吲哚-1,4-对苯醌衍生物。该方法具有条件温和、操作简单、试剂价廉等优点。部分化合物对人肺癌细胞系、人乳腺癌细胞和宫颈癌细胞系的抑制活性较好。有关化合物的进一步合成及深入的抗肿瘤活性研究正在进行中。
猜你喜欢
世界农药(2022年10期)2022-11-10
炼油技术与工程(2022年8期)2022-08-18
能源化工(2021年2期)2021-12-30
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
农药科学与管理(2021年2期)2021-03-16
昆明医科大学学报(2020年12期)2021-01-26
陶瓷学报(2020年6期)2021-01-26
人物画报(2020年29期)2020-03-14
人物画报(2020年36期)2020-03-13
世界农药(2019年3期)2019-09-10
