
大豆重组自交系群体异黄酮含量QTL连锁定位与关联定位的比较研究
2020-06-03 08:02刘再东孟珊贺建波邢光南王吴彬赵团结盖钧镒
中国农业科学 2020年9期
刘再东,孟珊,贺建波,邢光南,王吴彬,赵团结,盖钧镒
大豆重组自交系群体异黄酮含量QTL连锁定位与关联定位的比较研究
刘再东,孟珊,贺建波,邢光南,王吴彬,赵团结,盖钧镒
(南京农业大学大豆研究所/国家大豆改良中心/农业部大豆生物学与遗传育种重点实验室/作物遗传与种质创新国家重点实验室/江苏省现代作物生产协同创新中心,南京 210095)
【】异黄酮是大豆等豆类植物中富含的一类次生代谢产物,对食品和保健产业有重要作用。大豆籽粒可分离出12种异黄酮组分,可归为三大类:大豆苷类异黄酮、染料木苷类异黄酮和黄豆苷类异黄酮。通过鉴定大豆籽粒异黄酮总含量及3个组分含量性状的加性及上位性QTL,进而全面解析其复杂的遗传构成。利用先进2号和赶泰2-2双亲衍生的大豆重组自交系群体NJRSXG,在5个环境下测定4个异黄酮含量性状:异黄酮总含量(total isoflavone content,SIFC)、大豆苷类异黄酮总含量(total daidzin group content,TDC)、染料木苷类异黄酮总含量(total genistin group content,TGC)和黄豆苷类异黄酮总含量(total glycitin group content,TGLC)。选用混合模型复合区间作图法(mixed-model-based composite interval mapping,MCIM)和限制性两阶段多位点全基因组关联分析方法(restricted two-stage multi-locus genome-wide association analysis,RTM-GWAS)进行异黄酮含量QTL检测。2个亲本在4个异黄酮含量性状上均存在较大差异,重组自交系群体异黄酮含量在高值、低值2个方向上均出现超亲分离,低值方向分离趋势强于高值方向。利用连锁定位MCIM方法共检测到4个异黄酮含量性状的19个加性QTL和16对上位性QTL,分布于15条染色体上。第14染色体重要标记区间GNE186b—Sat020内检测到3个新加性QTL:、和,且表型变异解释率最高。利用关联定位RTM-GWAS方法分别检测到4个异黄酮含量性状的51、66、42和36个关联标记位点,表型变异解释率为39.7%—52.5%,检测到的位点中覆盖了MCIM方法检测的19个加性QTL中的11个以及11个上位性QTL。候选基因分析分别在加性QTL区域和上位性QTL区域检测到93和100个候选基因,富集分析显示在第14染色体重要标记区间GNE186b—Satt020内,、和的功能与异黄酮代谢有关。连锁定位和关联定位2种方法结合能相对全面地检测异黄酮含量QTL。与连锁定位方法MCIM相比,关联定位方法RTM-GWAS检测的QTL更多,总遗传贡献率更高,但尚不能检测上位性QTL,2种方法定位结果可相互验证补充,大豆籽粒异黄酮含量由大量QTL/基因控制。
大豆;重组自交系群体;异黄酮;连锁作图;关联定位;QTL
0 引言
【研究意义】异黄酮是一类重要的次级代谢产物,在大豆和其他豆类植物中含量较为丰富。异黄酮作为一种抗菌剂可以防御病原体和害虫对植物的入侵[1-2],医学研究显示人体每天摄入异黄酮可以预防前列腺癌、乳腺癌和骨质疏松症等疾病[3-4]。据报道,与大豆籽粒蛋白质和油脂含量相比,异黄酮总含量较低,仅为416—6 808 μg·g-1[5]。因此,培育高异黄酮含量大豆品种有利于食品和保健产业。【前人研究进展】大豆籽粒中可分离出12种异黄酮组分,依据苯环配基差异可归为三大类:大豆苷类异黄酮、染料木苷类异黄酮和黄豆苷类异黄酮,三类异黄酮的含量比例通常保持在4﹕5﹕1[6]。每类异黄酮再结合葡萄糖、乙酰葡萄糖和丙二酰葡萄糖形成相应的糖苷[2],其中,葡萄糖苷和乙酰葡萄糖苷在高效液相色谱(high performance liquid chromatography,HPLC)分析中含量较低,通常认为它们是样品预处理期间丙二酰葡萄糖苷的降解产物[7]。三类异黄酮的功能也存在一定差异。大豆苷类异黄酮的代谢产物雌马酚可以治疗乳腺癌,染料木苷类异黄酮作为某种酶的诱导剂有助于治疗动脉硬化[8],黄豆苷类异黄酮可以抑制成骨细胞的增殖以治疗骨质疏松症[9-10]。目前,多数研究报道了大量大豆籽粒异黄酮含量QTL(quantitative trait locus)分布于所有大豆染色体上[11-18]。Liang等[11]利用包含474个家系的重组自交系群体,检测到6个异黄酮含量QTL,分布在大豆4条染色体上。Wang等[13]利用重组自交系群体检测到34个异黄酮含量QTL,其中23个QTL为新发现位点,在Satt186与Satt226间的QTL在4个环境中均被检测到,表型变异解释率为3.4%—11.0%。Zhang等[16]检测到22个异黄酮含量QTL,分布在大豆8条染色体上,表型变异解释率为4.5%—7.9%,同时检测到3对异黄酮含量上位性QTL,其中,和影响黄豆黄苷含量,影响大豆异黄酮总含量。Zeng等[19]使用单因素方差分析和按性状分型的方法同时分析了加性以及加性与环境互作QTL。Gutierrez-Gonzalez等[20-22]使用混合模型复合区间作图法(MCIM)鉴定了一些在不同环境中均能稳定影响异黄酮含量的主效QTL。Li等[23]基于大豆重组自交系群体检测到48个加性QTL,表型变异解释率为3.0%—29.8%,其中,与黄豆苷类、染料木苷类和总异黄酮含量有关的8个QTL均被定位于A1连锁群的相同区域内,且表型变异解释率均大于14.0%,并经不同群体定位分析得以验证。Pei等[24]利用包含3 541个SLAF标记的高密度遗传图谱,在4个环境中检测到24个异黄酮含量QTL,其中,包括与染料木苷、丙二酰大豆苷、丙二酰染料木苷和总异黄酮含量相关的各1个新的QTL,另外还检测到9个上位性QTL。Cai等[25]通过构建高密度遗传图谱,于多环境下检测到15个异黄酮含量QTL,分布在大豆10条染色体上。异黄酮含量是由许多微效QTL控制的复杂数量性状,其中大部分QTL难以定位到遗传连锁图谱上,此外异黄酮含量在不同环境中的表型变异也是不稳定的[18-20]。目前,关于异黄酮相关基因、与异黄酮合成关键酶相互作用的转录因子的研究已有不少报道[26-28],朱莹等[26]克隆了一个与大豆异黄酮合成相关的R2R3类型MYB转录因子GmMYB184,验证了该转录因子对异黄酮合成途径关键基因的转录激活活性及其在异黄酮合成中的正向调控作用。Chu等[27]利用关联分析方法检测到与异黄酮含量显著相关的28个SNP标记及一个候选基因,并证明通过激活(异黄酮合酶2)和(查尔酮合成酶8)启动子,进而调控异黄酮含量。Vadivel等[28]研究表明转录因子GmMYB176通过激活的表达进而调控异黄酮的合成。【本研究切入点】全面解析异黄酮含量的遗传结构有助于了解大豆籽粒异黄酮含量的遗传模式并培育高异黄酮含量大豆新品种。获得可靠稳定的QTL是分子标记辅助育种的第一个重要步骤,由于不同的QTL检测方法对异黄酮含量的遗传解析效果不尽相同,因此,有必要比较不同定位方法以便使用更高效的统计模型在多种环境中检测异黄酮含量QTL。连锁定位MCIM方法基于多环境表型数据,不仅可以检测加性QTL和上位性QTL,还能够检测与环境互作QTL[29]。Pan等[30]基于大豆重组自交系群体全基因组SNP分子标记和开花期数据,比较了不同QTL定位方法和不同标记类型在QTL定位中的应用效果。结果显示基于SNP连锁不平衡区块(SNP linkage disequilibrium block,SNPLDB)标记的限制性两阶段多位点全基因组关联分析(restricted two-stage multi- locus genome-wide association analysis,RTM-GWAS)方法[31-32]不仅能检测较多的QTL,而且能合理估计QTL表型变异解释率,更适用于重组自交系群体。因此,连锁定位MCIM方法和关联定位RTM-GWAS方法的联合分析可能为全面解析异黄酮含量QTL提供新思路。【拟解决的关键问题】本研究在5个环境中,同时采用连锁定位MCIM方法和关联定位RTM-GWAS方法解析重组自交系群体NJRSXG异黄酮总含量(total isoflavone content,SIFC)、大豆苷类异黄酮总含量(total daidzin group content,TDC),染料木苷类异黄酮总含量(total genistin group content,TGC)和黄豆苷类异黄酮总含量(total glycitin group content,TGLC)4个异黄酮含量性状的遗传结构,并鉴定其加性QTL、上位性QTL以及与环境互作QTL。
1 材料与方法
1.1 材料与田间试验
以先进2号(异黄酮含量高)和赶泰-2-2(异黄酮含量低)为亲本,采用单粒传法衍生了包含147个家系的重组自交系群体NJRSXG。到2009年种植时,该群体处于F2:8:14世代。
供试材料为NJRSXG群体及其亲本,田间试验分别于2009、2010和2011年夏季在南京农业大学江浦农学试验站进行(长江以北),其中2009年夏季同时在南京农业大学溧水大豆试验点进行(长江以南)。另外,2010年在江浦同时进行了晚播试验,播种日期比同年正常日期推迟一个月,一些非生物胁迫如光照、时间和土壤水分在正常播种和晚播材料之间存在显著差异。因此,涉及5个环境,试验设计均采用完全随机区组设计,2次重复,穴播,每穴作为一个小区,每穴定苗8株,穴距0.8 m。每个小区内随机挑选100粒干燥成熟种子用于异黄酮含量测定。
1.2 异黄酮含量测定
参考王春娥等[33]方法进行异黄酮提取与含量测定。使用FOSS 1095 Knifetec研磨器将每份种子样品磨成细粉。样品分为2份,一份用于含水量测定(105℃,≥5 h);另一份用于提取异黄酮。将15 mg样品于2 mL微型离心管中,加入1.5 mL 80%(v/v)甲醇,50℃超声辅助提取1 h,冷却至室温后,将提取物以12 000 r/min离心10 min。将上清液通过Whatman 0.45-μm 25-mm PTFE有机相针式滤器过滤,取10 μL滤液使用Agilent Zorbax SB-C18色谱柱(4.6 mm×150 mm, 5 μm)通过Agilent 1100反相HPLC进行分析,柱温为50℃,流速为2.0 mL·min-1。流动相A为0.1%(v/v)乙酸,流动相B为100%甲醇。梯度洗脱为:0—2 min(27%B),2—3 min(27%—36%B),3—6 min(36%B),6—7min(36%—33%B),7—13 min(33%B),13—14 min(33%—27%B)和14—15 min(27%B)。检测波长为254 nm(DAD检测器,Agilent,美国)。
采用外标法定量异黄酮,使用12种异黄酮标准品。以每克干大豆籽粒异黄酮含量为指标(单位为μg·g-1)。共定量了4个异黄酮含量性状,分别为异黄酮总含量(SIFC)、大豆苷类异黄酮总含量(TDC)、染料木苷类异黄酮总含量(TGC)和黄豆苷类异黄酮总含量(TGLC)。SIFC为TDC、TGC和TGLC之和。TDC为大豆苷元(Daidzein)、大豆苷(Daidzin)、乙酰基大豆苷(6’-O-acetyldaidzin)和丙二酰基大豆苷(6’-O-malonyldaidzin)含量之和。TGC为染料木苷元(Genistein)、染料木苷(Genistin)、乙酰基染料木苷(6’-O-acetylgenistin)和丙二酰基染料木苷(6’-O-malonylgenistin)含量之和。TGLC为黄豆苷元(Glycitein)、黄豆苷(Glycitin)、乙酰基黄豆苷(6’-O-acetylglycitin)和丙二酰基(6’-O- malonylglycitin)黄豆苷含量之和。
1.3 遗传连锁图谱构建
NJRSXG群体遗传连锁图由国家大豆改良中心和中国科学院遗传与发育生物研究所共同构建[34]。使用来自SoyBase(http://soybase.org)的937个SSR标记,以及设计的540个EST-SSR和231个BAC-SSR,共1 732个分子标记筛选多态性,获得447个差异标记位点。最后使用JoinMap软件[35]将400个标记(285个SSR、74个EST-SSR和68个BAC-SSR)整合到一张连锁图谱中。遗传连锁图谱由23个连锁群组成,全长1 447.9 cM,标记平均间距为3.9 cM,标记顺序与大豆公共遗传连锁图谱[36]基本一致。
1.4 统计分析
根据如下线性模型进行多环境联合方差分析:
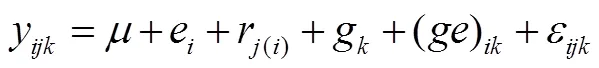
其中,为群体平均数,e为第个环境效应,r(i)为第个环境内第个区组的效应,g为第个基因型效应,()为基因型与环境互作效应,ε为随机误差。使用SAS软件[37]的PROC GLM进行联合方差分析,其中,环境、区组(环境)、基因型、基因型与环境互作,均视为随机效应。
1.5 QTL定位
采用混合线性模型复合区间作图法(MCIM)进行加性QTL、上位性QTL以及与环境互作加性和上位性QTL检测。利用QTLNetwork软件进行MCIM方法计算,以排列测验1 000次的值为统计检验阈值检测QTL,基因组扫描窗口大小为10 cM,步长为1 cM,其他参数保持默认。同时,还采用He等[31-32]发展的限制性两阶段多位点全基因组关联分析(RTM-GWAS)方法,检测异黄酮含量关联标记位点,QTL检测显著水平设0.01。RTM-GWAS方法基于多位点模型,同时能够检测与环境互作QTL。比较来自不同作图方法的结果时,若不同方法定位到的某两QTL间的遗传距离小于5 cM,则认为此两QTL是相同的,该位点被不同方法同时检测到[38-40]。
2 结果
2.1 异黄酮组分性状的表型变异
次数分布表及频率分布图显示4个异黄酮含量性状呈连续性变化,为数量性状特征(表1和图1)。NJRSXG群体2个亲本间存在显著差异,母本先进2号是异黄酮含量极高的大豆栽培品种,异黄酮总含量(SIFC)为6 302 μg·g-1,而父本赶泰-2-2的异黄酮总含量仅为3 401 μg·g-1。整体来看,群体在低值、高值2个方向上都出现了超亲分离,4个异黄酮含量性状低值方向的超亲分离趋势均强于高值方向(其中TGLC在高值方向未发现有超亲分离现象)。多环境联合方差分析显示,家系间异黄酮含量性状变异远大于家系与环境互作的变异,即使后者差异显著,但值不大,表明这些异黄酮含量性状在不同环境中表现相对一致。异黄酮含量性状遗传率极高,异黄酮总含量(SIFC)的遗传率为94.1%,大豆苷类异黄酮总含量(TDC)、染料木苷类异黄酮总含量(TGC)和黄豆苷类异黄酮总含量(TGLC)的遗传率为92.7%—95.4%。

表1 异黄酮含量(102 μg·g-1)的次数分布
SIFC:异黄酮总含量;TDC:大豆苷类异黄酮总含量;TGC:染料木苷类异黄酮总含量;TGLC:黄豆苷类异黄酮总含量;N:家系数;P1:先进2号;P2:赶泰-2-2;GCV:遗传变异系数;2:广义遗传率。下同
SIFC: total isoflavone content; TDC: total daidzin group content; TGC: total genistin group content; TGLC: total glycitin group content; N: the number of lines; P1: Xianjin 2; P2: Gantai-2-2; GCV: genetic coefficient of variation;2: broad sense heritability. The same as below
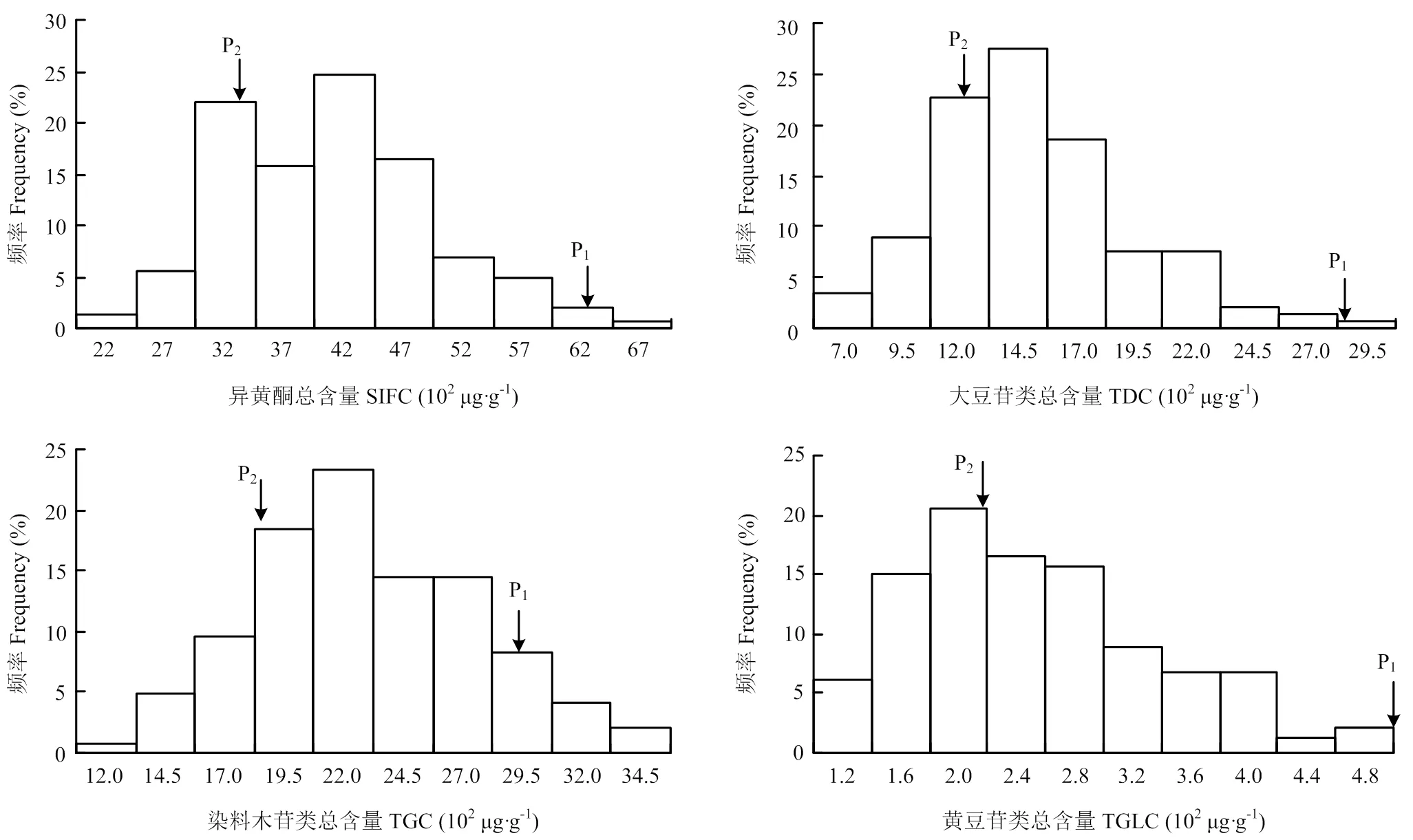
SIFC:异黄酮总含量;TDC:大豆苷类异黄酮总含量;TGC:染料木苷类异黄酮总含量;TGLC:黄豆苷类异黄酮总含量。下同
异黄酮含量性状间具有不同程度相关性(表2)。4个异黄酮组分性状,异黄酮总含量(SIFC)、大豆苷类异黄酮总含量(TDC)、染料木苷类异黄酮总含量(TGC)和黄豆苷类异黄酮总含量(TGLC)之间均呈正相关,SIFC与TDC、TGC相关系数分别为0.95和0.97,而与TGLC之间的相关性略低,相关系数为0.39。TDC、TGC和TGLC之间的相关性相对较低,相关系数分别为0.26和0.32,表明大豆苷类和染料木苷类异黄酮的生物合成和遗传变异相似,这两种成分对异黄酮总含量有显著影响,而黄豆苷类异黄酮的合成和变异有其自身的特点,这也暗示具有高度显著相关性的不同性状的一些QTL可能共享相同的基因组区域。
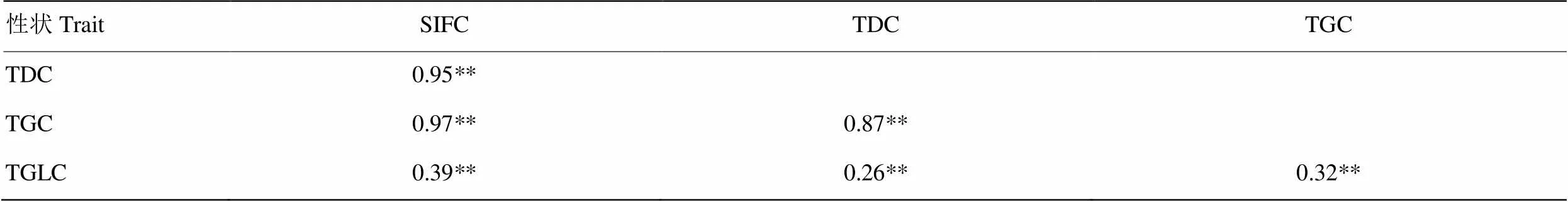
表2 异黄酮含量性状间相关系数
SIFC:异黄酮总含量;TDC:大豆苷类异黄酮总含量;TGC:染料木苷类异黄酮总含量;TGLC:黄豆苷类异黄酮总含量。**表示0.01显著性水平测验显著。下同
SIFC: total isoflavone content; TDC: total daidzin group content; TGC: total genistin group content; TGLC: total glycitin group content. ** represents significance at level of 0.01. The same as below
2.2 异黄酮含量的连锁定位
MCIM方法共检测到4个异黄酮总含量(SIFC)加性QTL和15个异黄酮组分含量(TDC、TGC和TGLC)的加性QTL(表3)。加性效应的总贡献率为15.9%(TGLC)—27.3%(TGC),上位性效应的总贡献率为1.0%(SIFC)—15.4%(TGLC)。而性状遗传率远大于其加性和上位性QTL的总贡献率,表明许多未被定位到的QTL贡献了大部分遗传率。单个QTL的表型变异解释率为0.2%—10.6%,其中和的贡献率均超过10.0%,并且被定位在相同标记区间GNE186b—Satt020内(表4)。此外,MCIM方法检测到与SIFC相关的1对上位性QTL,与其余3个组分含量性状相关的共15对上位性QTL,其表型变异解释率为0.2%—4.6%。
表3 异黄酮含量MCIM方法遗传解析
Table 3 The genetic dissection of isoflavone content architecture
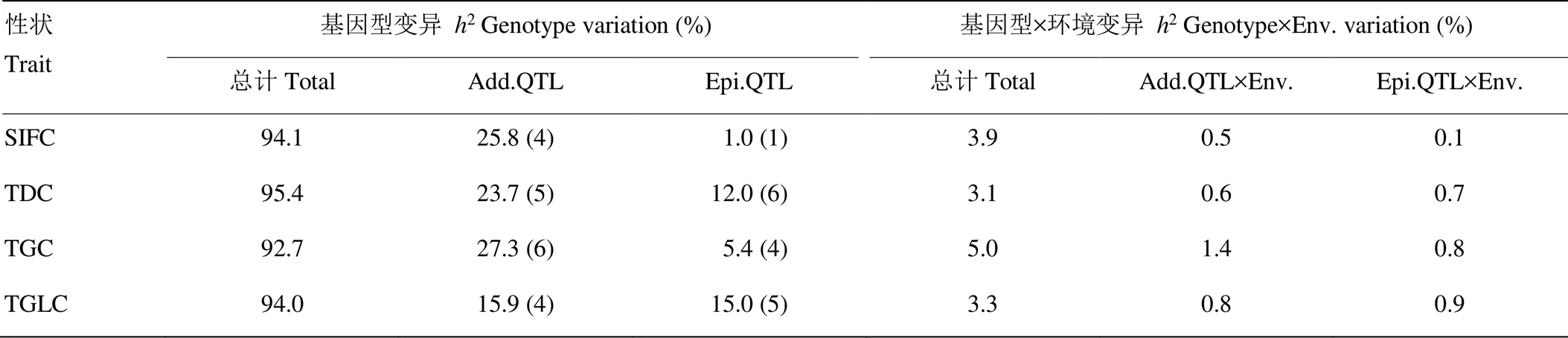
括号内数字为QTL个数或对数;2:遗传率;Add.:加性;Epi.:上位性
Number in parentheses is the number of QTLs or QTL pairs;2: heritability; Add.: additive; Epi.: epistatic
MCIM方法检测到与SIFC相关的4个加性QTL,分布在第3、14、18和19染色体上,QTL表型贡献率为4.2%—10.6%,合计25.8%(表4)。其中,的贡献率在5种环境中均超过10.0%。仅发现1对上位性QTL,表型贡献率为1.0%。上位性QTL的贡献率仅占SIFC遗传率(94.1%)的微小部分,未检测到的QTL总贡献率约为67.3%。检测到与TDC相关的5个加性QTL,分布于第3、8、14和18染色体上,QTL表型贡献率为2.3%—8.7%,合计23.7%。对每个单环境进行分析,也检测到了,表型贡献率为8.7%。共检测到6对上位性QTL,分布于9条染色体上,总贡献率为12.4%。加性QTL的总贡献率几乎是上位性QTL的2倍。检测到的QTL总贡献率为36.1%,未检测到的QTL解释了超过一半的表型变异。与TGC相关的6个加性QTL分布在第3、13、14、18和19染色体上,表型贡献率为0.2%—10.3%,总计27.3%。其中,的贡献率为10.3%,且在5种环境中都发现了该位点。同时检测到4对上位性QTL,分布在6条染色体上,总贡献率为5.4%。加性QTL的总贡献率远大于上位性QTL,两者累积为32.7%,未检测到的QTL解释了约60.0%的表型变异。检测到与TGLC相关的4个加性QTL,分布在第6、8、10和17染色体上。表型贡献率为2.9%—5.3%,总计15.9%。在7条染色体上检测到5对上位性QTL,总贡献率为15.4%。加性效应的总贡献率与上位性效应几乎相同,2种贡献率的总和为31.3%,小于未被定位的QTL总贡献率。
图2显示了异黄酮总含量(SIFC)及3个组分总含量(TDC、TGC和TGLC)性状中重叠的QTL或基因组区域。第3染色体上的Satt521—GNE324、第8染色体上的Satt089—Satt525、第14染色体上的GNE186b—Satt020、第18染色体上的Satt038—Sat_168及第19染色体上的Satt481—GNE091区间是主要的加性QTL区域,每个区域均包含不同性状的QTL,与SIFC相关的4个加性QTL全部位于主要区域内。第14染色体上的GNE186b—Sat020区域最为重要,位于该区域的3个加性QTL,、和在对应性状中均具有最高的贡献率,3个QTL的加性效应均为负,表明低异黄酮含量亲本赶泰-2-2为提高SIFC,TDC和TGC含量各提供了一个等位基因。而第1染色体上的Sat_353—Satt532b、第10染色体上的GNB134—Sat_282和GNE061—GNB130、第12染色体上的GNE229—Satt192、第14染色体上的Satt304—Sat_355和第17染色体上的Sat_022—Satt186标记区间是主要上位性QTL区域,每个区域内主要包括SIFC和TGC的上位性QTL,或者TDC和TGLC的上位性QTL。值得注意的是在11个主要QTL区域,除加性QTL区域内同时具备上位性外,其余区域内的QTL只有加性或上位性效应。
加粗位点表示同时为RTM-GWAS方法所检测到。2Add:加性QTL表型变异解释率;2Epi:上位性QTL表型变异解释率
Locus in bold style is also detected by RTM-GWAS.2Add: phenotypic variation explained by additive QTL;2Epi: phenotypic variation explained by epistatic QTL

环形代表主要的加性QTL区域,三角形代表主要的上位性QTL区域,连接2个QTL的直线代表上位性效应
2.3 异黄酮含量多位点关联分析
利用限制性两阶段多位点全基因组关联分析(RTM-GWAS)方法分别检测到51、66、42和36个与异黄酮总含量(SIFC)、大豆苷类异黄酮总含量(TDC)、染料木苷类异黄酮总含量(TGC)和黄豆苷类异黄酮总含量(TGLC)关联的标记位点(图3),总表型变异解释率(包括位点主效和位点与环境互作效应)分别为44.4%、52.5%、39.7%和45.6%(表5),其中位点主效表型变异解释率为34.9%(TGC)—47.0%(TDC),位点与环境互作效应表型变异解释率为2.3%(TGLC)—5.5%(TDC)。可见,异黄酮含量主要由主效位点控制,与环境互作效应相对较小,这与MCIM方法结果一致。
RTM-GWAS方法检测的51个异黄酮总含量(SIFC)位点中,45个位点主效显著,21个位点与环境互作效应显著,17个位点2种效应皆显著。51个位点覆盖了MCIM方法检测的4个加性QTL中的3个,分别为、和,覆盖率为75%。然而,这3个QTL在RTM-GWAS中的贡献率相对较小,其中对应的Satt556标记位点表型变异解释率最高,也仅为1.8%,但在MCIM中其解释率高达10.6%。而RTM-GWAS方法检测结果中第13染色体上标记位点Satt030的表型变异解释率最高,为6.3%。此外,RTM-GWAS方法检测的位点还覆盖了MCIM方法检测的一个上位性QTL。与大豆苷类异黄酮总含量(TDC)关联的66个位点中,60个位点主效显著,30个位点与环境互作效应显著,28个位点2种效应皆显著。66个位点覆盖了MCIM方法检测的5个加性QTL中的4个,以及4个上位性QTL,加性QTL的覆盖率高达80%。标记位点GNE186b的表型变异解释率最高,为8.6%,该位点对应的加性效应遗传率也最高,为8.7%。然而加性效应遗传率也较高为5.8%,但是与其对应的标记位点Sat_210表型变异解释率仅为0.4%。与染料木苷类异黄酮总含量(TGC)关联的42个位点中,40个位点主效显著,18个位点与环境互作效应显著,16个位点两种效应皆显著。42个位点仅覆盖了MCIM方法检测的6个加性QTL中的2个,分别为标记位点Satt521对应的和Satt020对应的。的加性效应遗传率最高,为10.3%,而标记位点的Satt020的表型变异解释率相对较低,为2.8%。另外,RTM-GWAS方法检测的位点还覆盖了MCIM方法检测的两个上位性QTL。与黄豆苷类异黄酮总含量(TGLC)关联的36个位点主效全部显著,其中有7个位点与环境互作效应显著。36个位点覆盖了MCIM方法检测的4个加性QTL中的2个,分别为标记位点GNB215a对应的和Sat_140对应的。的加性效应遗传率最高,为5.3%,与其对应的标记位点GNB215a的表型变异解释率也最高,为5.0%。Sat_140的表型变异解释率也较高,为3.5%。另外,RTM-GWAS方法检测的位点还覆盖了MCIM方法检测的4个上位性QTL。
2.4 大豆籽粒异黄酮含量候选基因
异黄酮含量的主要候选基因可以根据11个主要加性和上位性QTL区域推测。最终在主要加性QTL区域和主要上位性QTL区域分别检测到93和100个候选基因。基因富集分析结果显示这193个候选基因涉及21个生物学过程,主要与运输、信号转导、碳水化合物代谢等过程相关,表明控制异黄酮含量的基因体系包含与多种生物代谢过程相关的一系列基因(图4)。值得注意的是,在最重要的第14染色体加性QTL区域GNE186b—Satt020,3个基因、和的功能与异黄酮代谢有关,其中参与了类黄酮生物合成过程。
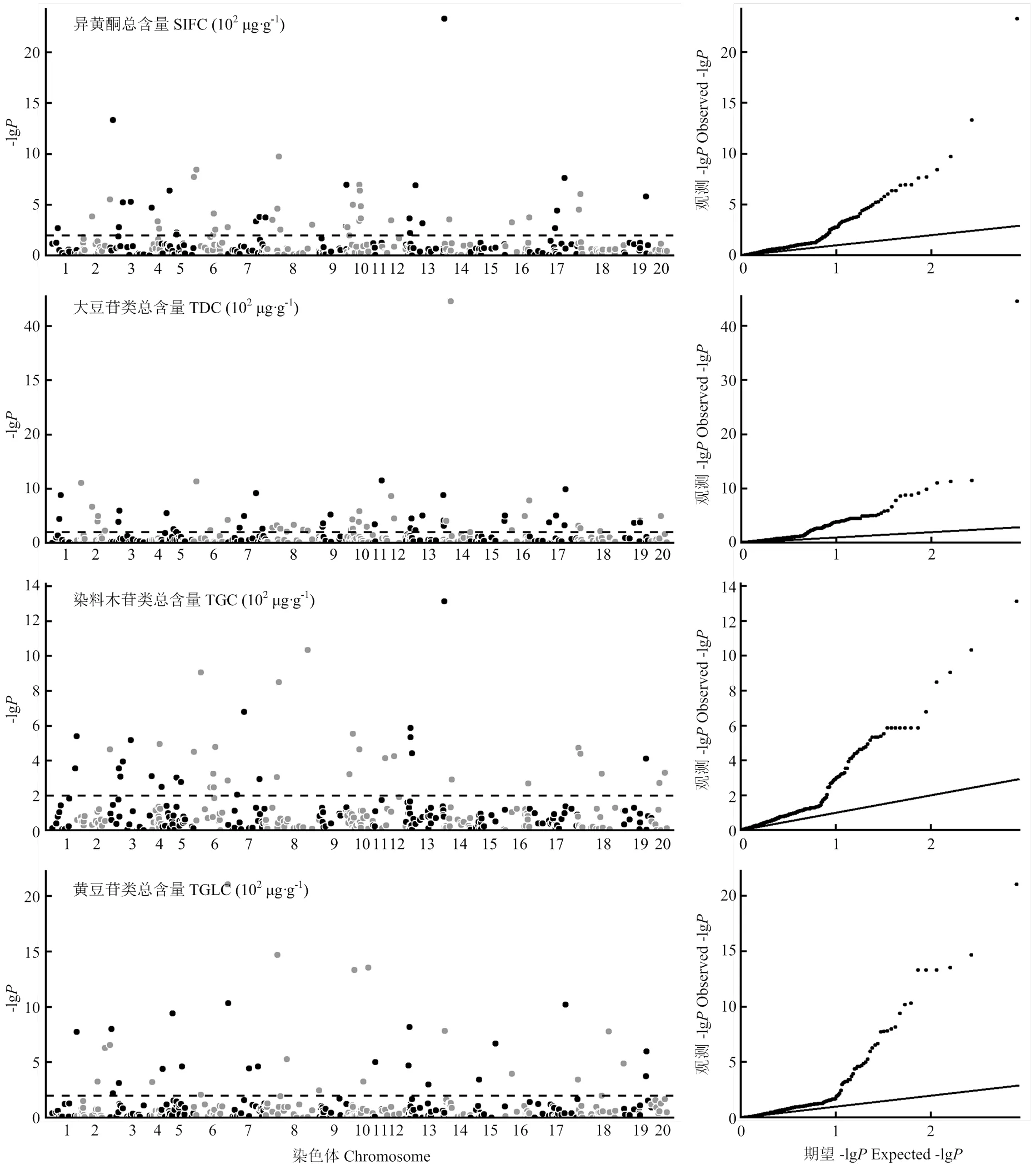
图3 异黄酮含量关联分析
3 讨论
3.1 与前人QTL定位结果的比较
整合在NJRSXG群体遗传图谱中的一些BAC- SSR标记不能锚定在大豆公共连锁图谱上,因此,必须使用前人和本研究中共有的标记来比较结果,当2个位点的物理距离小于2 Mb时,该位点被认为是相同的QTL。本研究19个异黄酮含量QTL中,9个QTL为新位点。其中黄豆苷类异黄酮总含量(TGLC)QTL的标记区间为Satt079—GNB215a,而根据大豆的公共遗传图谱[36],在Gutierrez-Gonzalez等研究中[21],的左侧标记Satt319与Satt079相距约4 cM。Kassem等[41]在第8染色体上鉴定到的QTL位点,位于Sat_129(位置,84.08 cM)和Satt097(122.05 cM)内,而本研究中的右侧标记Satt525(96.97cM)位于该区间内。Gutierrez-Gonzalez等[22]报道了第17染色体上,在Satt186和Sat_086(118.66 cM)之间定位的QTL位点,而在本研究中检测到的的标记区间Satt397(69.30 cM)—Satt_220(128.73 cM)覆盖,该位点具有候选基因的一个拷贝[21]。Yang等[14]在第3染色体上定位到SIFC的一个QTL位点,其临近的标记Satt521与本研究中的左侧标记相同,而另一个位于第12染色体上的QTL位点与本研究检测到的处于相同的标记区间内。根据大豆公共图谱,Yoshikawa等[15]定位到一个SIFC的QTL位点,与其相邻的标记Satt066位置是78.8 cM,而的右侧标记Satt020(72.10 cM)与其相距约6 cM,其中,与TGLC相关的一个QTL位点与本研究中的位置重叠,而与其定位到的相距较远,为2个不同的位点。此外,Gutierrez-Gonzalez等[20-22]和Zeng等[19]也检测到异黄酮含量的上位性QTL,本研究16对上位性QTL中,无任意1对与前人结果一致。然而其中10个上位性QTL(如和)的位置与前人定位的加性QTL位置一致(表4),这是否具有实际意义还需要进一步验证。
表5 异黄酮含量关联标记/QTL
Table 5 Marker/QTL detected for isoflavone component traits
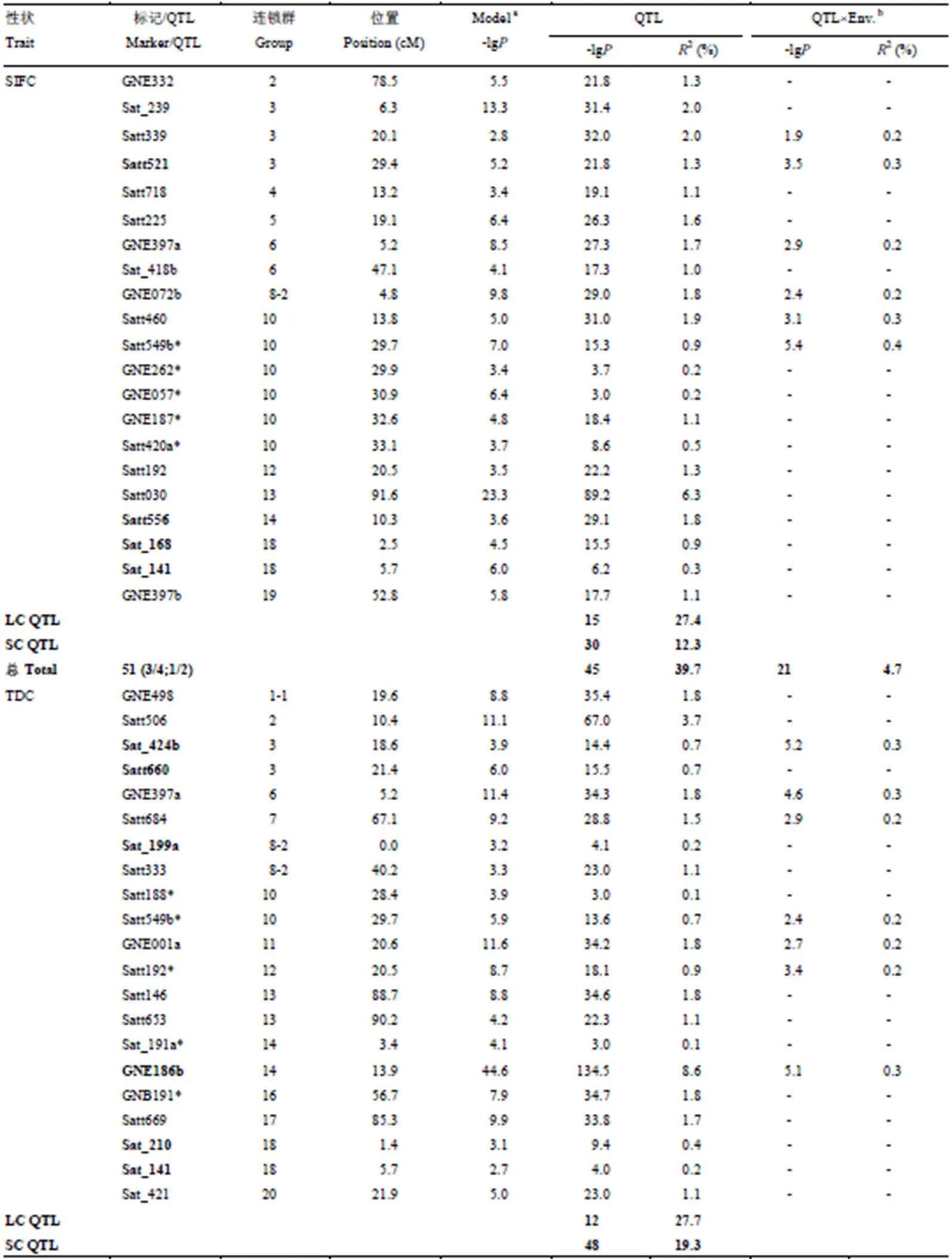
仅列出大效应位点以及与MCIM方法重叠的位点,加粗位点表示同时为MCIM方法所检测到加性位点,*表示同时为MCIM所检测到的上位性位点。标记/QTL列总和数分别表示RTM-GWAS方法检测的位点总数、覆盖MCIM方法所检测到的加性和上位性位点数目及其总数,例如51 (3/4;1/2)表示RTM-GWAS方法共检测到51个位点,覆盖MCIM方法所检测的4个加性位点中的3个,2个上位性位点中的1个。-lg列的总和数为总位点数。LC和SC QTL分别表示大贡献(2≥1%)和小贡献(2<1%)QTL;2:表型变异解释率;a:QTL检测模型显著性测验;b:QTL与互作效应
Only loci of large contribution or overlapping with MCIM result were showed. Locus in bold style is also detected as additive QTL by MCIM, * is also detected as epistatic QTL by MCIM. The total number in marker/QTL column represent the number of loci detected by RTM-GWAS, and the number of additive and epistatic QTL overlapped with MCIM, for example, 51 (3/4; 1/2) means a total of 51 loci are detected by RTM-GWAS covering 3 additive and 1 epistatic QTLs detected by MCIM, while 4 and 2 are the total number of additive and epistatic QTL detected by MCIM. Total numbers in column -lgare the number of loci. LC and SC QTL represent large (2≥1%) and small (2<1%) contribution QTL;2: phenotypic variance explained;a: statistical hypothesis testing performed in QTL detection model;b: QTL-by-environment interaction effect
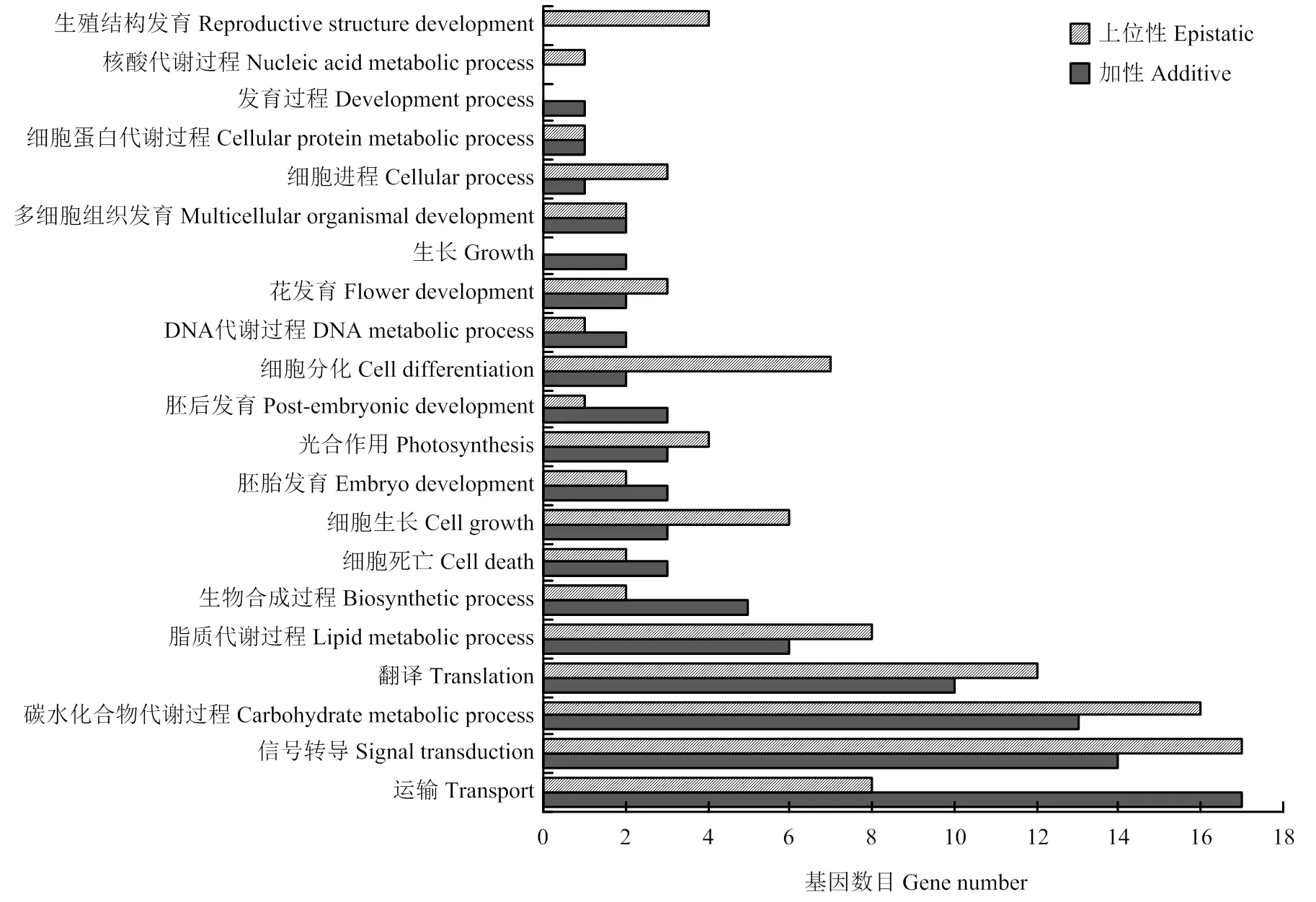
图4 大豆籽粒异黄酮含量候选基因生物学过程分布
3.2 异黄酮含量QTL体系
本研究中异黄酮含量的遗传方差由广义遗传率(来自方差分析)估计,方差来源于3部分,加性QTL效应,上位性QTL效应和未被定位的QTL效应。未被定位到的QTL的存在有如下原因:首先,未定位到的QTL确实为一些效应非常小的微效QTL,这种假设符合前人提出的“数量性状的主基因加多基因模型”[42]。另一方面,MCIM方法未定位到的QTL可能在其他作图群体或NJRSXG的改良群体中被检测到。因此,遗传学家必须使用具有较大样本量和高密度分子标记的不同群体来鉴定可靠的QTL,并以多种策略将连锁分析和关联分析相结合[43-45]。
4个异黄酮含量性状未定位到的总QTL解释率为59.3%—67.3%,占一半以上的遗传变异,这表明未被定位到的QTL在异黄酮含量的遗传中起主导作用,这将会给育种实践带来一定困难。但是,2个新的加性QTL(和)解释了超过相应性状10.0%的遗传变异,且还位于相同区域,该区域具有候选基因、和,且该主要遗传区域经关联分析研究得到验证,它可为异黄酮总含量(SIFC)和染料木异黄酮总含量(TGC)的分子标记辅助育种提供重要信息。此外,异黄酮总含量(SIFC)、大豆苷类异黄酮总含量(TDC)和染料木异黄酮总含量(TGC)的加性效应都远大于对应的上位性效应,而在黄豆苷类异黄酮总含量(TGLC)中,加性与上位性效应相当,表明黄豆苷类异黄酮的生物合成依赖于复杂的代谢网络,其合成途径在很大程度上仍然是未知的[46]。
异黄酮是源自苯丙烷类和类黄酮途径的次级代谢产物,可被包括非生物和生物胁迫在内的多种环境因素诱导[46],因此有必要设计多环境试验,从而经多环境联合分析以确定异黄酮的可靠遗传区域。除利用MCIM方法进行数据联合分析外,本研究还在5个环境下分别检测加性QTL(数据略),在19个加性QTL中,单个环境中共鉴定到了12个。第14染色体主要加性QTL区域GNE186b—Satt020上的3个QTL(、和),在5个环境中均被检测到,表明该遗传区域对SIFC、TDC和TGC的表型变异有稳定的遗传贡献。在4个、3个、2个和1个环境中检测到的QTL数量分别为0、3、1和5,说明这些QTL的效应可能是由某些特定的环境胁迫引起的。
目前,在大豆异黄酮基因的研究方面,已知不少与异黄酮合成有关的基因,应用分子生物学手段一些基因的功能已得到验证,本研究在第14染色体GNE186b—Satt020区域内定位到2个新的加性QTL(和),并预测同区域内的3个基因、和的功能与异黄酮代谢有关,其中参与了类黄酮生物合成过程,而关于这三个基因的详细功能仍是未知的,有待进一步研究。
3.3 连锁定位与关联定位的比较和互补
MCIM方法检测的位点往往也能为RTM-GWAS方法所检测到,位点表型贡献率与MCIM方法相当或相对较小,RTM-GWAS方法同时也检测到了其他大效应和小效应位点。MCIM方法共检测到与4个异黄酮含量性状(SIFC、TDC、TGC和TGLC)相关的6、17、13和12个QTL,而RTM-GWAS方法分别检测到51、66、42和36个与对应性状关联的标记位点。与连锁定位MCIM方法相比,关联定位RTM-GWAS方法能够检测到更多的异黄酮含量QTL,且覆盖了MCIM方法检测的19个加性QTL中的11个。MCIM方法检测的加性QTL总表型变异解释率较低,为15.9%—27.3%,而RTM-GWAS方法所检测位点的总表型变异解释率为39.7%—52.5%。
然而,RTM-GWAS方法尚不能检测上位性QTL,从MCIM方法检测的4个异黄酮含量性状的上位性QTL可以看出,异黄酮含量上位性QTL的效应较小,例如,异黄酮总含量(SIFC)上位性QTL贡献率仅为1.0%,染料木异黄酮总含量(TGC)上位性QTL贡献率为5.4%,并且异黄酮总含量(SIFC)、大豆苷类异黄酮总含量(TDC)和染料木异黄酮总含量(TGC)的加性效应均远大于上位性效应。因此,对于本研究的4个异黄酮含量性状,RTM-GWAS方法可以获得更为全面的遗传解析结果。
整合连锁定位与关联定位用于异黄酮含量的遗传解析,不仅利用连锁定位方法主效位点检测功效高的优点,而且还能利用RTM-GWAS方法位点检测功效高、遗传解析较为全面的优势,2种方法相互验证,从而对分析结果做出更加准确的推断。
4 结论
NJRSXG群体重组自交系中,异黄酮总含量(SIFC)、大豆苷类异黄酮总含量(TDC)、染料木异黄酮总含量(TGC)和黄豆苷类异黄酮总含量(TGLC)均出现超亲分离现象。利用混合模型复合区间作图法(MCIM)检测到了4个异黄酮含量性状的共19个加性QTL和16对上位性QTL,其中2个新发现的加性QTL解释了超过相应性状10.0%的遗传变异。利用限制性两阶段多位点全基因组关联分析方法(RTM-GWAS)检测到51、66、42和36个与SIFC、TDC、TGC和TGLC关联的标记位点,与4个异黄酮含量性状MCIM结果有4、8、4和6个位点存在重叠,位点覆盖率为25%—80%。连锁定位方法MCIM在主效位点的检测上具有较高的功效,而关联定位方法RTM-GWAS检测的QTL更多,总遗传贡献率更高,但尚不能检测上位性QTL,连锁定位和关联定位2种方法结合能相对全面地检测异黄酮含量QTL,2种方法定位结果可相互验证补充,从而对异黄酮含量遗传构成做出较为全面的推断。大豆籽粒异黄酮含量由大量QTL/基因控制。
[1] Graham T, Graham M, Subramanian S, Yu O. RNAi silencing of genes for elicitation or biosynthesis of 5-deoxyisoflavonoids suppresses race-specific resistance and hypersensitive cell death ininfected tissues., 2007, 144: 728-740.
[2] Griffith A P, Collison M W. Improved methods for the extraction and analysis of isoflavones from soy-containing foods and nutritional supplements by reversed-phase high-performance liquid chromatography and liquid chromatography-mass spectrometry., 2001, 913: 397-413.
[3] Munro I, Harwood M, Hlywka J, Stephen A, Doull J, Flamm W, Adlercreutz H. Soy isoflavones: a safety review., 2003, 61: 1-33.
[4] Tanaka M, Fujimoto K, Chihara Y, Torimoto K, Yoneda T, Tanaka N, Hirayama A, Miyanaga N, Akaza H, Hirao Y. Isoflavone supplements stimulated the production of serum equol and decreased the serum dihydrotestosterone levels in healthy male volunteers., 2009, 12: 247-252.
[5] 王春娥, 赵团结, 盖钧镒. 中国大豆资源异黄酮含量及其组分的遗传变异和演化特征. 中国农业科学, 2010, 43(19): 3919-3929.
Wang C E, Zhao T J, Gai J Y. Genetic variability and evolutionary peculiarity of isoflavone content and its components in soybean germplasm from China., 2010, 43(19): 3919-3929. (in Chinese)
[6] Wang H, Murphy P A. Isoflavone content in commercial soybean foods., 1994, 42: 1666-1673.
[7] Reinli K, Block G. Phytoestrogen content of foods: A compendium of literature values., 1996, 26: 123-148.
[8] Schrader C, Ernst I M, Sinnecker H, Soukup S, Kulling S E, Rimbach G. Genistein as a potential inducer of the anti-atherogenic enzyme paraoxonase-1: studies in cultured hepatocytes in vitro and in rat liver in vivo., 2012, 16(10): 2331-2341.
[9] Winzer M, Rauner M, Pietschmann P. Glycitein decreases the generation of murine osteoclasts and increases apoptosis., 2010, 160(17/18): 446-451.
[10] Yoshida H, Teramoto T, Ikeda K, Yamori Y. Glycitein effect on suppressing the proliferation and stimulating the differentiation of osteoblastic MC3T3-E1 cells., 2001, 5: 1211-1213.
[11] Liang H Z, Yu Y L, Wang S F, Lian Y, Wang T F, Wei Y L, Gong P T, Liu X Y, Fang X J, Zhang M C. QTL mapping of isoflavone, oil and protein contents in soybean (L. Merr.)., 2010, 9: 1108-1116.
[12] Wang Y, Han Y, Teng W, Zhao X, Li Y, Wu L, Li D, Li W. Expression quantitative trait loci infer the regulation of isoflavone accumulation in soybean (L. Merr.) seed., 2014, 15: 680.
[13] Wang Y, Han Y, Zhao X, Li Y, Teng W, Li D, Zhan Y, Li W. Mapping isoflavone QTL with main, epistatic and QTL × environment effects in recombinant inbred lines of soybean., 2015, 10: e0118447.
[14] Yang K, Moon J, Jeong N, Chun H, Kang S, Back K, Jeong S. Novel major quantitative trait loci regulating the content of isoflavone in soybean seeds., 2011, 33: 685-692.
[15] Yoshikawa T, Okumoto Y, Ogata D, Sayama T, Teraishi M, Terai M, Toda T, Yamada K, Yagasaki K, Yamada N, Tsukiyama T, Yamada T, Tanisaka T. Transgressive segregation of isoflavone contents under the control of four QTLs in a cross between distantly related soybean varieties., 2010, 60: 243-254.
[16] Zhang H J, Li J W, Liu Y J, Jiang W Z, Du X L, Li L, Li X W, Su L T, Wang Q Y, Wang Y. Quantitative trait loci analysis of individual and total isoflavone contents in soybean seeds., 2014, 93: 331-338.
[17] Akond M, Liu S, Kantartzi S K,KHALID M, NACER B, LIGHTFOOT D A, YUAN J Z, WANG D C, ANDERSON J, KASSEM M A. A SNP genetic linkage map based on the ‘Hamilton’ by ‘Spencer’ recombinant inbred line population identified QTL for seed isoflavone contents in soybean., 2015, 134(5): 580-588.
[18] PRIMOMO V S, POYSA V, ABLETT G R, JACKSON C J, GIJZEN M, RAJCAN I. Mapping QTL for individual and total isoflavone content in soybean seeds., 2005, 45: 2454-2464.
[19] Zeng G, Li D, Han Y, Teng W, Wang J, Qiu L, Li W. Identification of QTL underlying isoflavone contents in soybean seeds among multiple environments., 2009, 118: 1455-1463.
[20] Gutierrez-Gonzalez J J, Vuong T D, Zhong R, Yu O, Lee J D, Shannon G, Ellersieck M, Nguyen H T, Sleper D A. Major locus and other novel additive and epistatic loci involved in modulation of isoflavone concentration in soybean seeds., 2011, 123: 1375-1385.
[21] Gutierrez-Gonzalez J J, Wu X, Gillman J D, Lee J D, Zhong R, Yu O, Shannon G, Ellersieck M, Nguyen H T, Sleper D A. Intricate environment-modulated genetic networks control isoflavone accumulation in soybean seeds., 2010, 10: 105.
[22] Gutierrez-Gonzalez J J, Wu X, Zhang J, Lee J D, Ellersieck M, Shannon J G, Yu O, Nguyen H T, Sleper D A. Genetic control of soybean seed isoflavone content: importance of statistical model and epistasis in complex traits., 2009, 119: 1069-1083.
[23] Li X H, Kamala S, Tian R, hui d, li w l, kong y b, zhang c y. Identification and validation of quantitative trait loci controlling seed isoflavone content across multiple environments and backgrounds in soybean., 2018, 38(1): 8.
[24] Pei R, Zhang J, Tian L, zhang s g, han f x, yang s r, wang l z, li b, sun j m. Identification of novel QTL associated with soybean isoflavone content., 2018, 6(3): 244-252.
[25] Cai Z, Cheng Y, Ma Z, liu x g, ma q b, xia q j, zhang g y, mu y h, nian h. Fine-mapping of QTLs for individual and total isoflavone content in soybean (L.) using a high-density genetic map., 2018, 131(3): 555-568.
[26] 朱莹, 褚姗姗, 张培培, 程浩, 喻德跃, 王娇. R2R3-MYB转录因子GmMYB184调节大豆异黄酮合成. 作物学报, 2018, 44(2): 185-196.
Zhu y, chu s s, zhang p p, cheng h, yu d y, wang j. An R2R3-MYB transcription factor GmMYB184 regulates soybean isoflavone synthesis., 2018, 44(2): 185-196. (in Chinese)
[27] Chu S S, WANG J, Zhu Y, LIU S L, ZHOU X Q, ZHANG H, WANG C, TIAN Z, CHENG H, YU D Y. An R2R3-type MYB transcription factor, GmMYB29, regulates isoflavone biosynthesis in soybean., 2017, 13(5): e1006770.
[28] Vadivel A, Kumaran A, Kagale S, DHAUBHADEL S. GmMYB176 regulates multiple steps in isoflavonoid biosynthesis in soybean., 2019, 10: 562.
[29] Yang J, Zhu J, Williams R W. Mapping the genetic architecture of complex traits in experimental populations., 2007, 23: 1527-1536.
[30] Pan L y, He J b, Zhao T j, Xing G n, Wang Y f, Yu D y, Chen S y, Gai J y. Efficient QTL detection of flowering date in a soybean RIL population using the novel restricted two-stage multi-locus GWAS procedure., 2018, 131: 2581-2599.
[31] He J b, Meng S, TZhao t j, Xing g n, Yang S p, Li Y, Guan R z, Lu J j, Wang Y f, Xia Q j, Yang B, Gai J y. An innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding., 2017, 130: 2327-2343.
[32] 贺建波, 刘方东, 邢光南, 王吴彬, 赵团结, 管荣展, 盖钧镒. 限制性两阶段多位点全基因组关联分析方法的特点与计算程序. 作物学报, 2018, 44(9): 1274-1289.
HE J B, LIU F D, XING G N, WANG W B, ZHAO T J, GUAN R Z, GAI J Y. Characteristics and calculation procedures of a restrictive two-stage multi-site genome-wide association analysis method., 2018, 44(9): 1274-1289. (in Chinese)
[33] 王春娥, 赵团结, 盖钧镒. 大豆异黄酮组分HPLC快速分析技术及其在豆腐加工中的应用. 作物学报, 2010, 36(12): 2062-2072.
Wang C E, Zhao T J, Gai J Y. Establishment of a rapid HPLC method for quantifying isoflavone components and its application in tofu processing., 2010, 36(12): 2062-2072. (in Chinese)
[34] 王宇峰. 大豆基因组SSR分布特征和高密度遗传图谱的构建、整合与应用[D]. 南京: 南京农业大学, 2009.
Wang Y F. Genomic characterization of simple sequence repeats and establishment, integration and application of high density genetic linkage map in soybean[D]. Nanjing: Nanjing Agricultural University, 2009. (in Chinese)
[35] Stam P. Construction of integrated genetic linkage maps by means of a new computer package: Join Map., 1993, 3(5): 739-744.
[36] Song Q J, Marek L F, Shoemaker R C, Lark K G, Concibido V C, Delannay X, Specht J E, Cregan P B. A new integrated genetic linkage map of soybean., 2004, 109: 122-128.
[37] SAS Institute Inc. SAS/STAT User's Guide. Cary, NC: SAS Institute Inc, 2017.
[38] Chantret N, Mingeot D, Sourdille P, Bernard M, Jacquemin J M, Doussinault G. A major QTL for powdery mildew resistance is stable over time and at two development stages in winter wheat., 2001, 103: 962-971.
[39] Symonds V V, Godoy A V, Alconada T, Botto J F, Juenger T E, Casal J J, Lloyd A M. Mapping quantitative trait loci in multiple populations of Arabidopsis thaliana identifies natural allelic variation for trichome density., 2005, 169: 1649-1658.
[40] 邢光南, 周斌, 赵团结, 喻德跃, 邢邯, 陈受宜, 盖钧镒. 大豆抗筛豆龟蝽(Fabricius)的QTL分析. 作物学报, 2008, 34(3): 361-368.
Xing G N, Zhou B, Zhao T J, Yu D Y, Xing H, Chen S Y, Gai J Y. Mapping QTLs of resistance to(Fabricius) in soybean., 2008, 34(3): 361-368. (in Chinese)
[41] Kassem M A, Shultz J, Meksem K, Cho Y, Wood A J, Iqbal M J, Lightfoot D A. An updated ‘Essex’ by ‘Forrest’ linkage map and first composite interval map of QTL underlying six soybean traits., 2006, 113: 1015-1026.
[42] 盖钧镒, 章元明, 王健康. 植物数量性状遗传体系. 北京: 科学出版社, 2003.
Gai J Y, Zhang Y M, Wang J K.. Beijing: Science Press, 2003. (in Chinese)
[43] Famoso A N, Zhao K, Clark R T, Tung C W, Wright M H, Bustamante C, Kochian L V, McCouch S R. Genetic architecture of aluminum tolerance in rice () determined through genome-wide association analysis and QTL mapping., 2011,7: e1002221.
[44] Krill A M, Kirst M, Kochian L V, Buckler E S, Hoekenga O A. Association and linkage analysis of aluminum tolerance genes in maize., 2010, 5: e9958.
[45] 苏成付, 赵团结, 盖钧镒. 不同统计遗传模型QTL定位方法应用效果的模拟比较. 作物学报, 2010, 36(7): 1100-1107.
Su C F, Zhao T J, Gai J Y. Simulation comparisons of effectiveness among qtl mapping procedures of different statistical genetic models., 2010, 36(7): 1100-1107. (in Chinese)
[46] Yu O, McGonigle B. Metabolic engineering of isoflavone biosynthesis., 2005, 86: 147-190.
A comparative study on linkage and association QTL mapping for seed isoflavone contents in a recombinant inbred line population of soybean
LIU ZaiDong, MENG Shan, HE JianBo, XING GuangNan, WANG WuBin, ZHAO TuanJie, GAI JunYi
(Soybean Research Institute, Nanjing Agricultural University/National Center for Soybean Improvement/Key Laboratory of Biology and Genetic Improvement of Soybean (General), Ministry of Agriculture/State Key Laboratory for Crop Genetics and Germplasm Enhancement/Jiangsu Collaborative Innovation Center for Modern Crop Production, Nanjing 210095)
【】Isoflavones are a group of phenolic secondary metabolites which are relatively abundant in soybean and some other legumes, and are important for food and healthcare industry. A total of 12 kinds of components are isolated from soybean seed, and can be grouped into three categories: daidzin group, genistin group and glycitin group. To understand the complex genetic constitutions of isoflavone content in soybean, the additive and epistatic quantitative trait loci (QTLs) conferring the total isoflavone content and its component contents were detected in the present study. 【】The NJRSXG recombinant inbred line (RIL) population derived from Xianjin 2 and Gantai-2-2 were used in this study. Four isoflavone content traits, i.e. the total seed isoflavone content (SIFC), the total daidzin group content (TDC), the total genistin group content (TGC) and the total glycitin group content (TGLC) were tested in five environments. The mixed model composite interval mapping (MCIM) and restricted two-stage multi-locus genome-wide association analysis (RTM-GWAS) were used for QTL detection. 【】There was a large difference in isoflavone content between the two parental lines of NJRSXG population, and transgressive segregations were observed in NJRSXG population while the transgressive trend in low isoflavone content direction were stronger than that in high isoflavone content direction.A total of 19 additive QTLs and 16 pairs of epistatic QTLs for the four isoflavone traits on 15 chromosomes were detected by MCIM. Three novel additive QTLs, i.e.,and, were detected in the same important marker interval GNE186b-Sat020 on chromosome 14, and explained the highest phenotypic variation. A total of 51, 66, 42 and 36 significantly associated markers were detected by RTM-GWAS for SIFC, TDC, TGC and TGLC, respectively. The phenotypic variation explained by these markers was ranged from 39.7% to 52.5%, covering 11 additive QTLs and 11 epistatic QTLs detected by MCIM. Furthermore, a total of 93 and 100 candidate genes were annotated in the additive and epistatic QTL regions, respectively. Gene enrichment analysis indicated that three genes located in the important marker interval GNE186b-Satt020 on chromosome 14, i.e.,and, were related to isoflavone metabolism. 【】A relatively thorough detection of isoflavone content QTLs was achieved by using linkage and association mapping. Compared with the linkage mapping method MCIM, the association mapping method RTM-GWAS can detect more QTLs with larger total contribution to phenotypic variation, but cannot detect epistatic QTLs as in MCIM. The QTLs detected from the two methods can be used for complementary verification from each other.A large number of QTLs/genes are involved in the seed isoflavone contents of soybean.
soybean [(L.) Merr.]; isoflavone; genetic dissection; linkage mapping; association mapping; QTL

10.3864/j.issn.0578-1752.2020.09.006
2019-09-09;
2020-01-02
国家自然科学基金(31701447)、国家作物育种重点研发计划(2017YFD0101500,2017YFD0102002)、长江学者和创新团队发展计划(PCSIRT_17R55)、教育部111项目(B08025)、中央高校基本科研业务费项目(KYT201801)、农业部国家大豆产业技术体系CARS-04、江苏省优势学科建设工程专项、江苏省JCIC-MCP项目
刘再东,E-mail:2714699171@qq.com。孟珊,E-mail:mshan84@163.com。刘再东和孟珊为同等贡献作者。通信作者贺建波,E-mail:hjbxyz@gmail.com。通信作者盖钧镒,E-mail:sri@njau.edu.cn
(责任编辑 李莉)
猜你喜欢
中国现代医生(2022年21期)2022-08-22
天津医科大学学报(2021年1期)2021-01-26
食品安全导刊(2020年30期)2020-11-23
医药前沿(2020年20期)2020-11-10
三农资讯半月报(2020年2期)2020-03-09
中国食品(2018年7期)2018-09-10
车迷(2018年12期)2018-07-26
家庭医药·快乐养生(2017年4期)2017-04-19
山东工业技术(2016年15期)2016-12-01
癌变·畸变·突变(2015年3期)2015-02-27
