
东北大豆种质群体百粒重QTL-等位变异的全基因组解析
2020-06-03 08:02郝晓帅傅蒙蒙刘再东贺建波王燕平任海祥王德亮杨兴勇程延喜杜维广盖钧镒
中国农业科学 2020年9期
郝晓帅,傅蒙蒙,刘再东,贺建波,王燕平,任海祥,王德亮,杨兴勇,程延喜,杜维广,盖钧镒
东北大豆种质群体百粒重QTL-等位变异的全基因组解析
郝晓帅1,傅蒙蒙1,刘再东1,贺建波1,王燕平2,任海祥2,王德亮3,杨兴勇4,程延喜5,杜维广2,盖钧镒1
(1南京农业大学大豆研究所/国家大豆改良中心/农业部大豆生物学与遗传育种重点实验室/作物遗传与种质创新国家重点实验室/江苏省现代作物生产协同创新中心,南京 210095;2黑龙江省农业科学院牡丹江分院/国家大豆改良中心牡丹江试验站,黑龙江牡丹江 157041;3黑龙江省农垦科学院,黑龙江佳木斯 154007;4黑龙江省农业科学院克山分院,黑龙江克山 161606;5长春市农业科学院,长春 130111)
【】对东北大豆种质群体百粒重性状进行全基因组关联分析,全面解析中国大豆主产区百粒重QTL-等位变异遗传构成,为东北地区大豆籽粒大小遗传改良提供理论基础。以东北地区育种和生产上常用的290份大豆材料作为试验群体,于2013和2014年在东北第二生态亚区的克山、牡丹江、佳木斯和长春4个地点进行百粒重表型鉴定试验。利用RAD-seq方法对试验群体进行基因组测序分析,对原始SNP数据进行过滤及填补缺失数据后,最终获得了82 966个高质量的SNP标记。根据限制性两阶段多位点全基因组关联分析(restricted two-stage multi-locus genome-wide association analysis,RTM-GWAS)方法,首先构建获得15 546个具有复等位变异的SNPLDB标记,然后使用两阶段多位点模型对百粒重性状进行全基因组关联分析。对检测到的百粒重关联SNPLDB标记位点附近(50 kb范围内)的基因进行分析,根据基因内SNP与SNPLDB标记位点之间关联性的卡方测验,筛选可能与百粒重性状相关的候选基因并进行功能注释。最后基于检测的百粒重QTL-等位变异体系分析了不同熟期组材料间的遗传分化。试验群体百粒重变异范围为18.3—20.7 g,性状遗传率为92.3%。RTM-GWAS方法共检测到76个与大豆百粒重性状关联的SNPLDB标记位点,其中15个位点主效不显著,另外61个主效显著位点解释了65.40%的表型变异;68个与环境互作效应显著的位点解释了17.46%的表型变异,另外8个位点与环境互作效应不显著。在检测到的76个位点中有34个位点与已报道的30个百粒重QTL重叠,另外42个位点为本研究新检测百粒重位点。基于检测的SNPLDB标记位点,共筛选到137个百粒重相关候选基因,功能注释显示这些候选基因不仅参与大豆百粒重的调节,还参与了初级新陈代谢、蛋白质修饰、物质运输、胁迫响应和信号转导等。对各熟期组间QTL-等位变异的遗传分化分析显示,尽管熟期组间百粒重差异不明显,但其QTL-等位变异遗传结构却发生了新生和汰除的变化。RTM-GWAS方法能相对全面地解析东北大豆种质群体百粒重QTL-等位变异遗传构成。东北大豆种质群体百粒重由大量QTL调控,且QTL与环境互作效应大,QTL存在丰富的复等位变异。由RTM-GWAS方法建立的QTL-等位变异矩阵为群体遗传及演化研究提供了新工具。
大豆;百粒重;限制性两阶段多位点全基因组关联分析;QTL-allele矩阵;候选基因
0 引言
【研究意义】百粒重是大豆产量重要构成因素之一[1-3],并受多基因控制,有较高的遗传力[4-5]。全面解析大豆百粒重的遗传机制,并挖掘控制大豆百粒重基因对大豆高产育种具有重要意义。另外,生育期是大豆光周期反应重要生态指标,决定着大豆在不同纬度、地区的种植范围,对产量、品质和适应性都至关重要[6]。东北地域辽阔,大豆资源是美洲大豆的主要种质基础,研究东北大豆百粒重的遗传基础,对世界大豆育种具有重要意义。【前人研究进展】随着科技的发展和大豆公共数据的扩增,越来越多的科研工作者投入到了百粒重QTL(quantitative trait locus)定位和全基因组关联分析研究中[7-10]。目前,SoyBase (http://soybase.org)数据库已收录约280个基于连锁分析检测的大豆百粒重性状QTL,全基因组关联分析检测的百粒重相关位点也约有90个。例如,Sun等[11]通过构建重组自交家系定位到分布在5个连锁群体上23个大豆百粒重QTL。在所有定位到的QTL中,有9个通过复合区间作图法得到,另外14个通过多区间作图法得到的。Kastoori等[12]利用3个大豆重组自交家系作图群体,并利用这三个群体构建了联合连锁图谱,最终定位到1个百粒重相关主效QTL。该研究同时还鉴定到了一些百粒重候选基因,这些基因参与了蛋白转运、氨基酸合成等过程。Kato等[13]利用日本和美国2个不同遗传背景下的栽培种构建了2个重组自交家系,在3个环境下定位到了15个与大豆百粒重QTL。以上研究结果为从分子水平揭示大豆百粒重性状的遗传机制奠定了基础。虽然基于连锁分析的QTL定位方法可以估计QTL的位置和效应,但由于其通常仅涉及2个亲本,因此,该方法所能检测到的等位变异较少,例如在重组自交系群体中,每个位点最多有2个等位变异,所以连锁定位方法无法较全面的解析数量性状。基于自然群体的全基因组关联分析为全面解析数量性状提供了方法,其可以检测到群体内单个位点上所有等位变异,相比于连锁定位更加全面,定位精度也比较高。例如,Hao等[14]通过构建关联分析群体,并在5个环境下种植,结合1 142个单核苷酸多态性(single-nucleotide polymorphism,SNP)和209个单倍型进行关联分析,分别定位到40个和9个与大豆百粒重性状显著关联的SNP位点和单倍型。其中,可以同时在3个环境、4个环境以及5个环境下都检测到的SNP分别为3、2和4个。Zhou等[15]通过对302份大豆自然群体进行至少11×的重测序,利用全基因组关联分析的方法检测到第3、13和17染色体上共计4个大豆百粒重位点。Sonah等[16]对试验材料进行高密度测序,检测到第2、13和20染色体上的3个百粒重性状显著关联的区域。【本研究切入点】尽管全基因关联分析分析已广泛用于动植物数量性状遗传解析,然而以往方法主要基于双等位SNP标记进行分析[17-18],由于自然群体存在广泛的复等位变异,因此,SNP标记无法估计位点的复等位变异效应。其次,以往关联分析研究通常基于单位点模型,忽略了相邻位点间的相互作用[19],导致表型变异解释率可能溢出(>2,甚至>100%)。另外,单位点模型对每个位点的假设测验均相互独立,这会导致多重测验标准的设置问题,进而导致较高的全试验错误率。对此,以往方法通过提高显著水平进行多重测验矫正,例如Bonferroni方法[20-21],而这又导致以往方法仅能检测少数位点,进而导致遗传率缺失。针对上述全基因组关联分析在数量性状遗传解析中的限制,He等[22]通过构建具有复等位变异的SNPLDB(SNP linkage disequilibrium block)标记,并基于多位点复等位变异模型,提出了限制性两阶段多位点全基因组关联分析(restricted two-stage multi-locus genome-wide association analysis,RTM-GWAS)方法。该方法基于多位点模型,使用常规显著性水平0.01或0.05,无需进行额外多重测验矫正。多位点模型充分考虑了相邻位点间的相互影响,因此,所检测位点表型变异解释率不会超过性状遗传率。目前,该方法已应用于多个数量性状遗传解析研究[23-25]。东北是中国大豆的主产区[26],有着复杂多变的生态环境和相应的的生态类型。研究东北地区代表性品种群体百粒重的遗传结构可以为该地区百粒重乃至产量的育种改良提供参考。【拟解决的关键问题】本研究以东北地区290份大豆材料为试验群体,该群体不仅时间跨度大,类型多,而且包含了东北地区近100年来大豆育种的遗传变异。利用RTM-GWAS方法并结合该群体两年四点表型数据进行关联分析,并利用结果进行候选基因预测及不同大豆成熟期组间控制百粒重性状的遗传结构变化的研究,以期全面解析大豆百粒重性状的遗传机制,并为未来选育高产优质的大豆品种提供理论支撑。
1 材料与方法
1.1 材料与田间试验
以2010—2012年在东北地区收集到的在1916—2010年种植比较广泛的361份大豆品种为试验材料。该群体具有衍生后代多,高产,油脂含量高,抗病等特点。2013—2014年将该群体在包括克山(KS)、牡丹江(MDJ)、佳木斯(JMS)和长春(CC)4个代表性地点东北地区的第二生态亚区进行田间试验。采用重复内分组设计,穴播,小区面积为1 m2,每小区种植4穴,每穴保留4株植株,4次重复。待到初花时期,仅调查至少拥有2穴、每穴中至少3株的小区。各试验点采用常规田间管理。各试验点品种正常成熟后,将小区内植株混合收获,室内脱粒后,32℃烘干48 h,然后随机选取100粒种子称量3次取平均值。由于材料之间成熟期差异比较大,361份材料中的71份成熟期过长,最终没有获得百粒重的数据,因此表型数据实际为290份正常成熟材料(包括9份地方品种、276份育成品种以及5份国外品种,电子附表1)的百粒重数据。
1.2 线性模型与方差分析
试验数据采用多年多点随机区组方法做近似方差分析,SAS软件PROC GLM程序中方差分析的线性模型为:
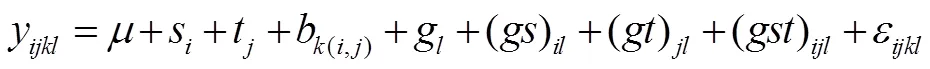
其中,y为第个年份第个地点下第个区组内第个品种的表型观测值,为群体平均数,s为第个年份效应,t为第个地点效应,b(i,j)为第个年份第个地点下第个区组的效应,g为第个品种的效应,()为第个年份与第个品种的互作效应,()为第个地点与第个品种的互作效应,()为第个年份、第个地点与第个品种的三级互作效应,ε为随机误差效应。品种视为固定效应,年份、地点、区组以及互作效应视为随机效应。
1.3 全基因组SNP分析
使用RAD-seq(restriction-site-association DNA sequencing)对290份材料在深圳华大基因进行简化测序。采用常规的CTAB法从新鲜大豆幼苗叶片中提取DNA,借助Illumina Hiseq 2000测序平台并结合多元鸟枪法进行基因组分析[27]。利用SOAP2软件[28]并参考大豆参考基因组Wm82.a1.v1.1[29]对测序所获得的序列进行比对。利用RealSFS检测SNP位点,之后对检测到的SNP位点按照缺失和杂合率≤20%和最小等位基因频率(MAF)≥1%的标准过滤[30],并利用fastPHASE[31]软件对缺失数据填补,最终获得82 966个高质量的SNP。
1.4 全基因组关联分析
根据He等[22]提出的限制性两阶段多位点全基因组关联分析(RTM-GWAS)方法,首先基于全基因组SNP构建获得了15 546个具有复等位变异的SNPLDB标记,每个SNPLDB标记的等位变异数目变化范围为2—9个。然后基于全基因组SNPLDB标记计算个体间的遗传相似系数,并提取特征向量用于控制全基因组关联分析的群体结构。最后,利用多位点模型对百粒重性状进行全基因组关联分析,显著水平设为0.05。由于多位点模型内建全试验误差控制,因此无需进行额外的多重测验矫正。以上计算分析采用RTM- GWAS软件[22]完成。
同时,基于SNPLDB构建的遗传相似系数矩阵,使用MEGA 7.0软件[32]构建了Neighbor-joining聚类树以观察群体结构是否异常。
1.5 候选基因的预测
根据检测到的QTL预测候选基因的方法,首先将定位到的SNPLDB两端各扩展50 kb,然后根据SoyBase(http://soybase.org)上提供的基因信息,将全部落在扩展后的SNPLDB区间内的基因选出。然后对每一个选出的基因中的全部SNP和SNPLDB之间的关联进行卡方(Chi-square)检验,显著性水平设为0.05。
2 结果
2.1 东北大豆种质群体百粒重的表型和基因型变异特征
各试验点百粒重性状平均值的次数分布和描述统计见表1。东北地区290份大豆品种百粒重平均值为19.8 g,变幅为8.2—29.5 g。不同环境下百粒重平均数变幅为18.3—20.7 g,百粒重最小为6.4—9.7 g,最大为28.2—32.0 g,环境间百粒重存在较大差异。
东北地区大豆百粒重两年四点联合方差分析显示(表2),百粒重在品种间有极显著差异,基因型、年份、地点间两两互作以及三级互作效应也呈现极显著,说明百粒重存在基因型与环境互作效应。但相比基因型方差,互作效应方差相对较小,多地点百粒重遗传率为0.923,单地点下遗传率变幅为0.642—0.780。
2.2 东北大豆种质群体百粒重全基因组关联分析
聚类分析(图1-a)显示东北大豆种质群体具有一定的群体结构,但群体分化相对不明显,基于SNPLDB标记的主成分分析(图1-b)也显示该群体虽然有一定的分类倾向,但整体上没有明显的聚类特征。
使用RTM-GWAS方法,第一阶段筛选出12 305个候选标记,第二阶段最终检测到76个与大豆百粒重性状显著关联的SNPLDB标记,分布在大豆18条染色体上(表3、图1-c和图1-d),每条染色体上检测到2—7个不等,其中第15、17和20染色体上最少,均只检测到2个SNPLDB标记,第6和18染色体上最多,均检测到7个显著关联的SNPLDB标记。第5和11染色体上没有检测到与大豆百粒重性状相关的SNPLDB标记。由于RTM-GWAS基于多位点模型,所有QTL在同一模型进行拟合,因此,一个位点只能筛选到一个显著的标记,而且通过对存在QTL比较多的染色体上的位点之间物理距离比较发现,相邻2个QTL之间的距离最小为0.58 Mb,最大达到18.21 Mb,且绝大多数相邻位点间的物理距离都超过了5 Mb,因此QTL在各条染色体上并非成簇分布。关联的76个位点中,有15个位点主效不显著,8个位点与环境互作效应不显著。61个主效显著位点总表型变异解释率为65.40%,68个位点与环境互作效应显著位点总表型变异解释率为17.46%,合计解释了82.86%的表型变异(表3)。61个主效显著位点包括18个大效应(2≥1%)位点和43个小效应(2<1%)位点,分别解释了52.15%和13.25%的表型变异。与以往研究比较显示,检测的76个SNPLDB标记中,有34个与前人报道的30个QTL存在重叠,另外42个SNPLDB位点为本研究新检测到的位点。
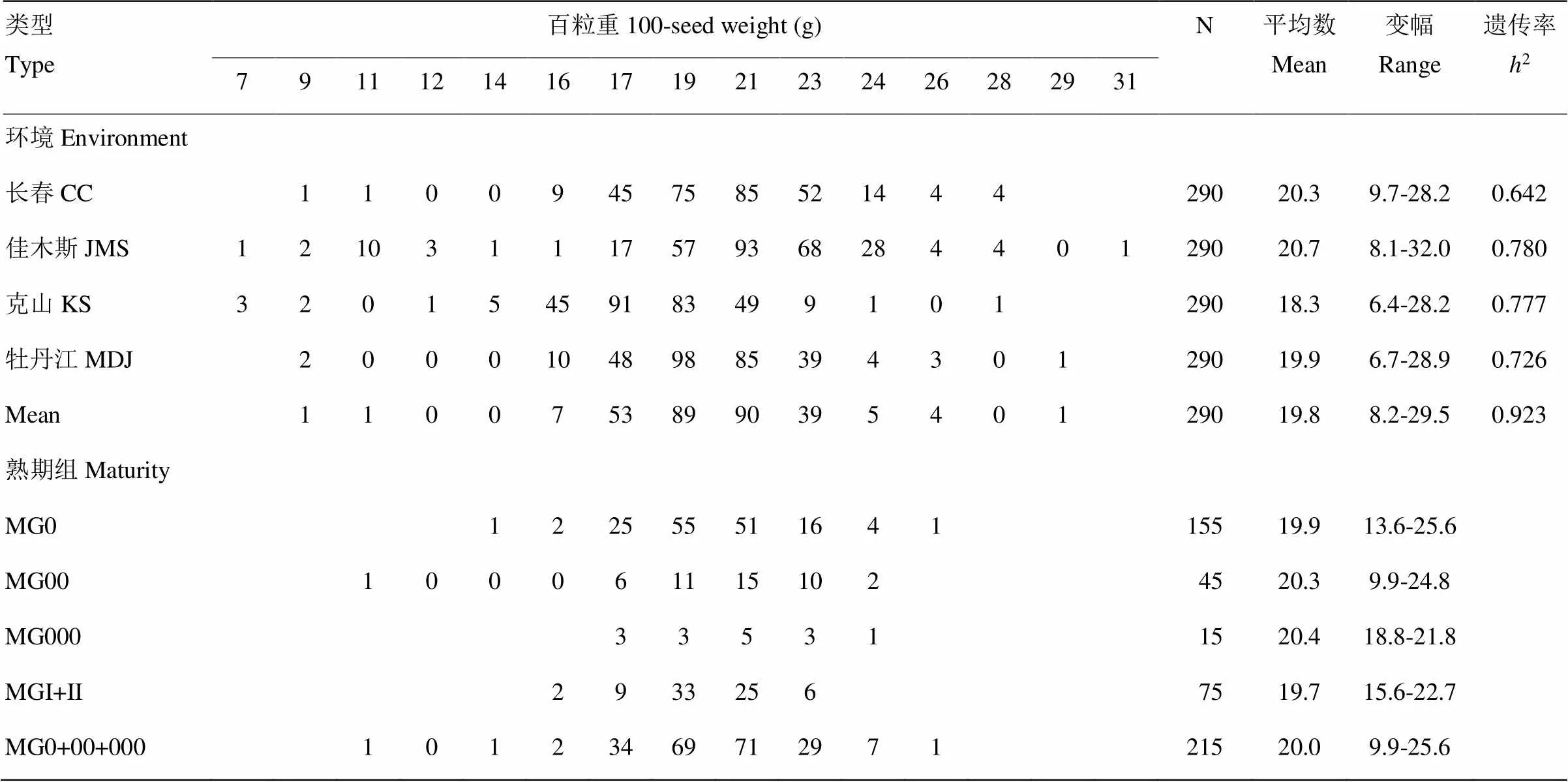
表1 东北大豆种质群体百粒重次数分布及描述统计
MG0、MG00、MG000分别是3个早期成熟期组的名称;MG0+00+000是MG0、MG00、MG000的合并名称;MGI+II是MGI和MGII的合并名称
MG0, MG00, MG000 are three early maturity groups; MG0+00+000 is the union of MG0, MG00 and MG000; MGI+II is the union of MGI and MGII
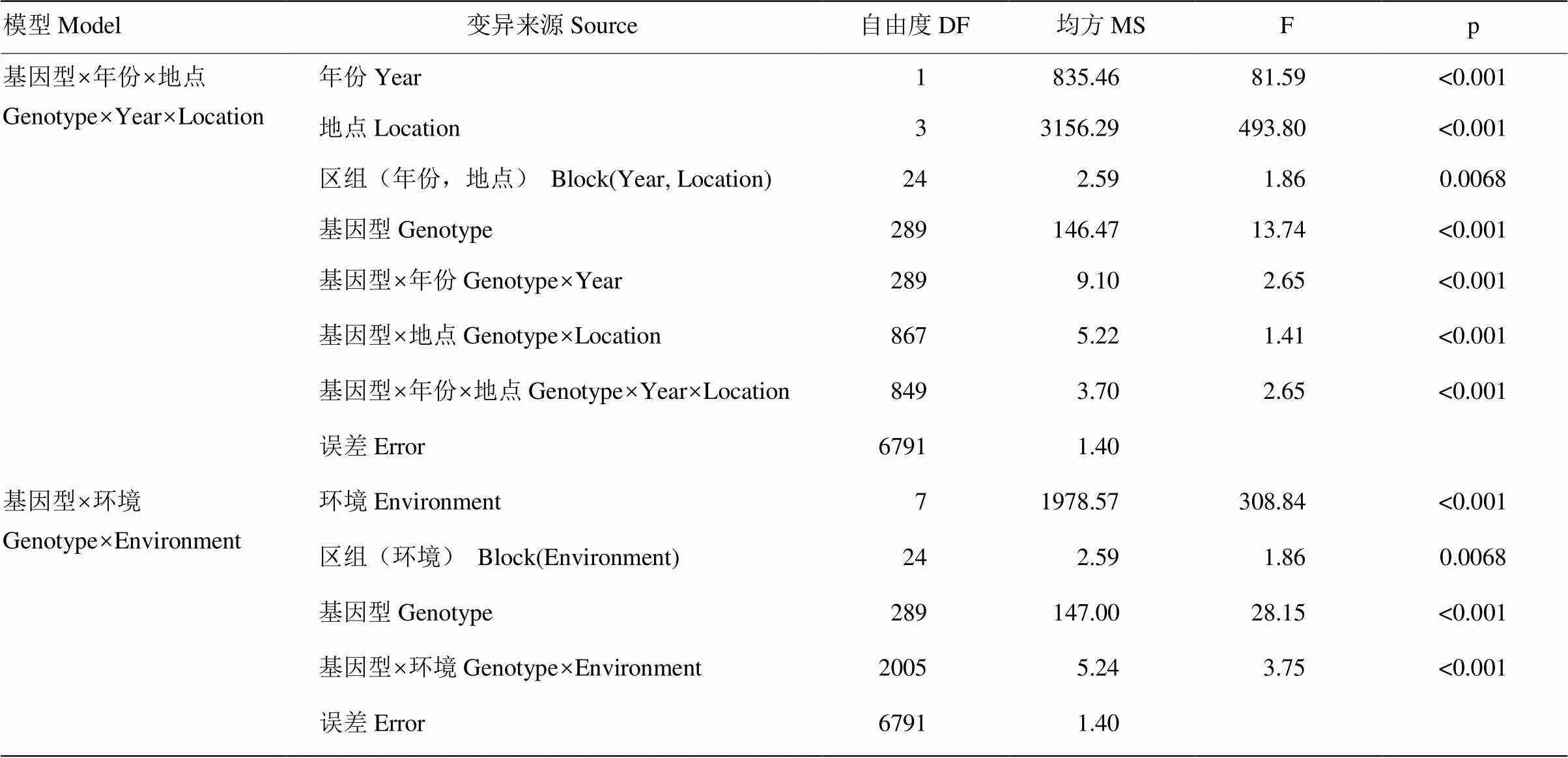
表2 东北大豆种质群体百粒重多年多点联合方差分析
基因型×环境模型为年份和环境合并为环境后的方差分析,用于RTM-GWAS关联分析
In Genotype×Environment model, Year and Location are combined into Environment which is used in RTM-GWAS
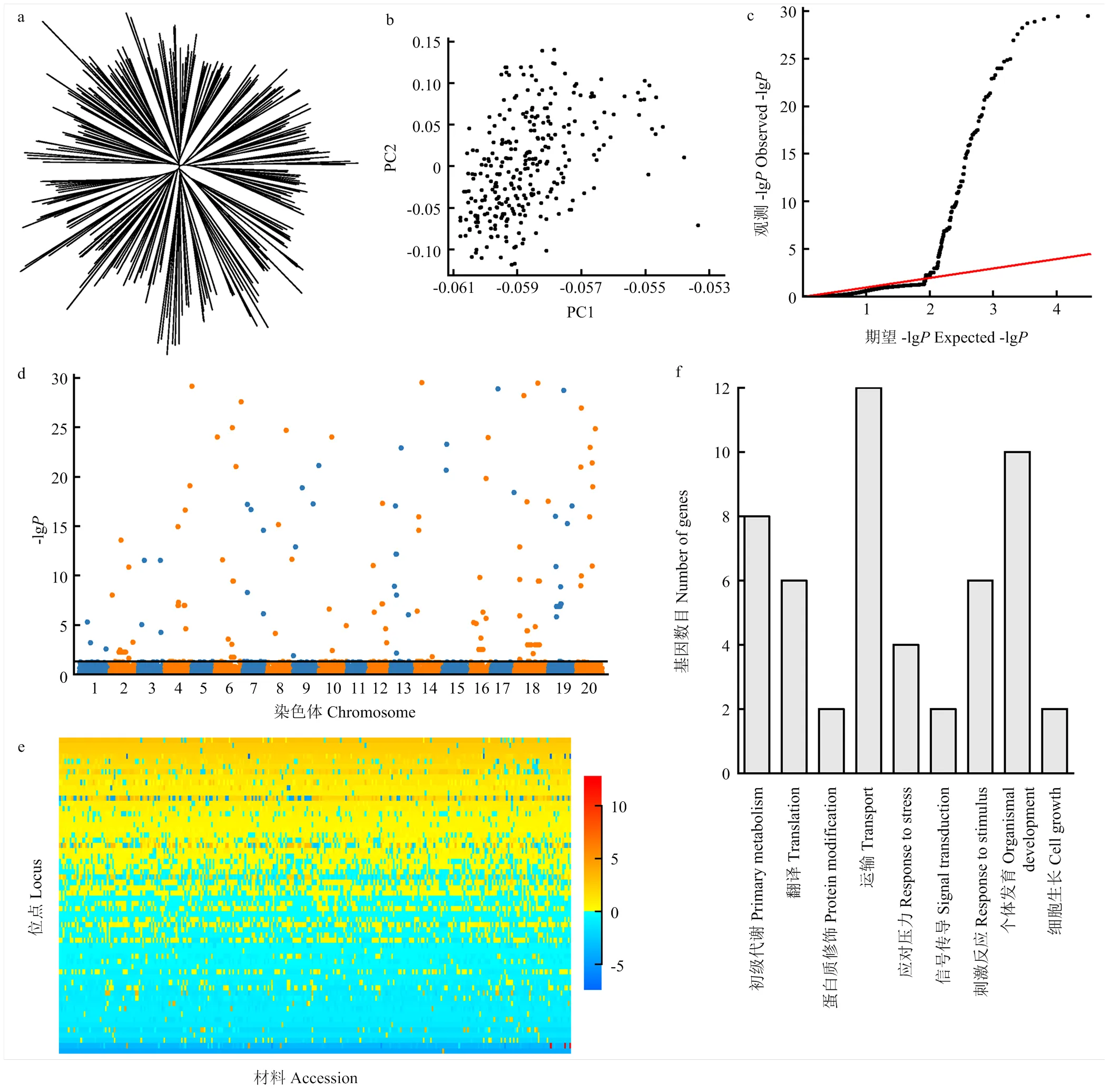
a:neighbor-joining聚类树;b:遗传相似系数矩阵特征向量散点图,PC1、PC2分别表示前2个特征向量;c:RTM-GWAS方法QQ图。其中-lgP大于30的记为30;d:RTM-GWAS方法Manhattan图;e:东北大豆种质群体百粒重QTL-allele矩阵;f:百粒重候选基因GO生物过程分布
表3 大豆百粒重显著相关SNPLDB位点
Table 3 SNPLDBs significantly associated with 100-seed weight in soybean
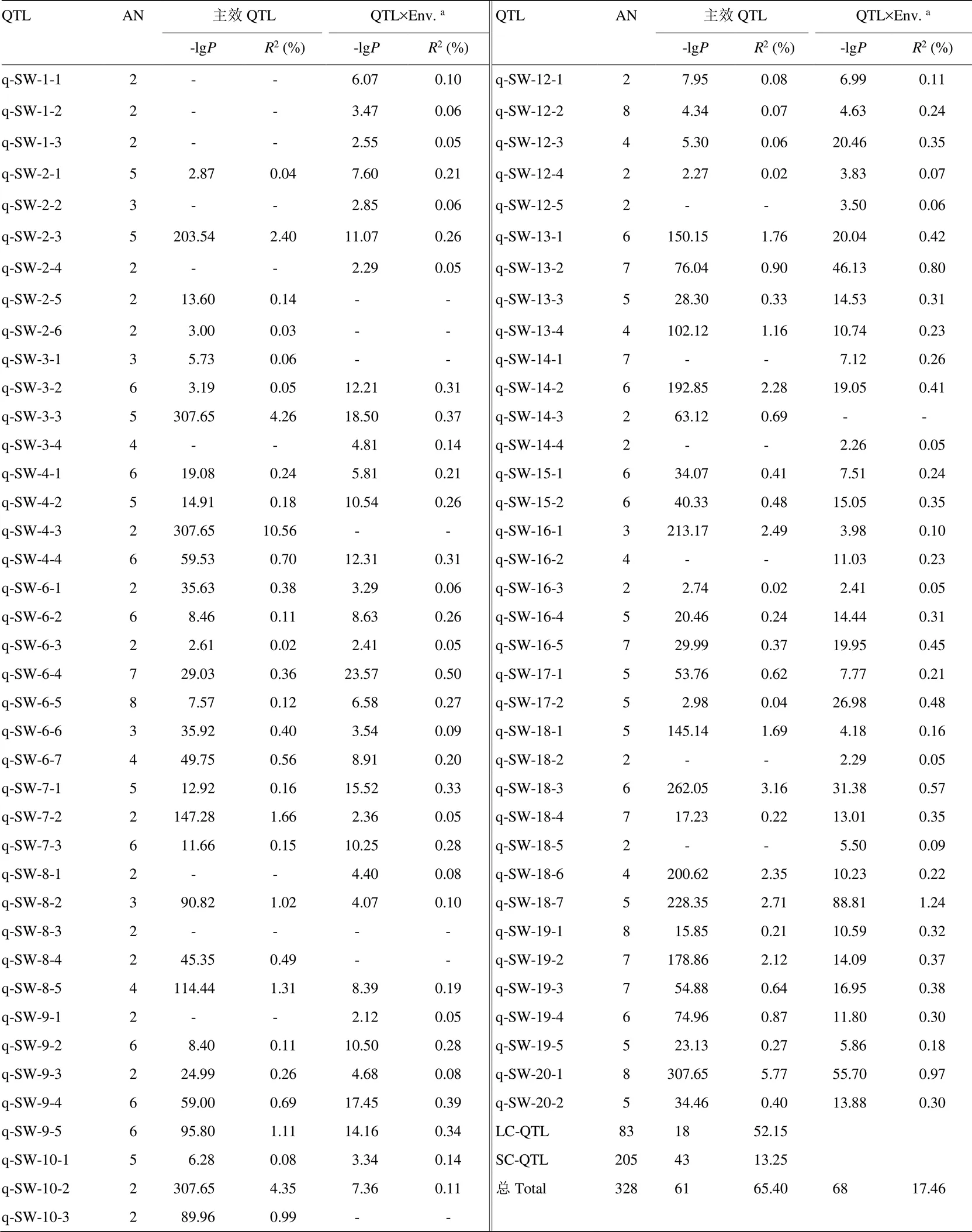
AN:等位变异数目。a:QTL与环境互作效应。LC-QTL和SC-QTL分别为大贡献(2≥1%)和小贡献(2<1%)QTL
AN: number of alleles.a: QTL-by-environment interaction effect. LC-QTL and SC-QTL represent large (2≥1%) and small (2<1%) contribution QTL
2.3 东北大豆种质群体百粒重QTL-allele矩阵及候选基因
与大豆百粒重关联的61个主效显著位点等位变异数目为2—8个,共计288个,其中47个位点存在复等位变异。等位变异效应值变化范围是-7.39—12.74,并进一步构建了61×290(位点×材料)的QTL-allele矩阵(图1-e)。该矩阵代表了东北大豆种质群体百粒重性状的遗传构成,可进一步用于群体分化、候选基因分析以及优化组合设计。
61个主效显著位点中仅有39个位点上或其扩展区域中存在共计739个基因,其中602个基因中没有SNP或者没有检测到与SNPLDB显著连锁的SNP,另外137个基因中包含了248个与SNPLDB显著关联的SNP。GO分析显示这137个基因中的83个涉及多种生物过程,包括初级新陈代谢、翻译、蛋白修饰、物质运输、胁迫响应、信号转导,对刺激的响应、器官发育、细胞生长以及一些未知过程(图1-f)。例如,候选基因内包含了8个SNP,组成了7种单倍型,对应着位点中的5种等位变异。由于候选基因前3个SNP并不在位点的区间内,因此,候选基因的单倍型数目比位点的等位变异数目多。另外,12个大效应QTL筛选到的候选基因主要参与大豆的代谢、转录、翻译、对刺激响应以及一些未知的生物过程,6个大效应QTL位点没有筛选到候选基因(表4)。
2.4 东北大豆种质群体新熟期组百粒重QTL-等位变异的新生和汰除
将大豆品种按照生育期进行熟期组划分,有助于品种交流、引种以及育种方案的设计。根据盖钧镒等[33]提出的中国大豆熟期组划分方法,将本研究群体材料划分为000、00、0、Ⅰ和Ⅱ等5个熟期组。其中,Ⅰ和Ⅱ为东北大豆种质旧熟期组,000、00和0为东北大豆种质新熟期组。不同熟期组的百粒重均值变幅为19.7—20.4 g,虽然平均数差异不大,但百粒重最小值为9.9—18.8 g,最大值为21.8—25.6 g,各熟期组之间百粒重变幅差异较大(表1)。基于百粒重QTL-等位变异体系分析,东北大豆熟期缩短过程中百粒重位点等位变异发生了一定的新生和汰除(表5)。这里将旧熟期组Ⅰ和Ⅱ合并(MGI+Ⅱ)作为基础,将新熟期组遗传结构与之比较,结果显示,从MGI+Ⅱ到MG0,MG0中同时新生了一些大效应的正效应和负效应等位变异,例如在QTL上新生了第1、2、7和8号等位变异,QTL的上新生了第1号等位变异。的第8号和QTL的第1号等位变异分别是本研究中百粒重QTL-等位变异体系中正负效应的最大值。由MG0到MG00,MG0中新生的QTL的第8号等位变异以及其他33个小到中等正效应的等位变异在MG00中被汰除,但MG00中汰除的负效应大部分为小效应等位变异。由MG00到MG000,MG000汰除了一批效应较大的负效和正效等位变异,其中就包括最大负效应等位变异(的第1号等位变异,效应值为-7.39)以及较大正效等位变异(的第7号等位变异,效应值为4.74)。而新增的等位变异中最大正、负效应分别仅为1.20和-3.78,这就使得MG000中材料的百粒重变幅在2个方向均大幅缩小,即从9.9—24.8到18.8—21.8。
进一步将熟期组0、00与000的材料合并(MG0+00+000)后与MGI+Ⅱ进行比较分析。从熟期组MGI+Ⅱ到0,到00,再到000的过程中,新生的等位变异累计共56个(34+3+19),但MGI+Ⅱ和MG0+00+000相差的新生等位变异却只有36个(表5)。同时从熟期组MGI+Ⅱ到0,到00,再到000的过程中,汰除的等位变异共计140个(5+74+61),但MGI+Ⅱ和MG0+00+000相差的汰除等位变异只有4个,这说明一些等位变异在MG0、MG00、MG000之间存在新生和汰除的反复过程,即在一个熟期组中被汰除后,又在其他熟期组中作为新生等位变异重新出现。例如,QTL上的第1号等位变异在MG0中被汰除后,又在MG00新生出来,并继续传递给了MG000,QTL的第8号等位变异在MG0组中新生,随后又在MG00中被汰除。综上所述,等位变异在不同熟期组间正效和负效等位变异几乎是同等数量新生或汰除,使得东北大豆群体各熟期组之间百粒重均值变化不大。但各熟期组的QTL-等位变异构成不尽相同,从而导致各熟期组之间百粒重的变化范围呈现差异。因而新生的熟期组群体中,百粒重的QTL-等位变异结构是有差异的,说明百粒重还有重组的潜力。
3 讨论
本研究采用限制性两阶段多位点全基因组关联分析(RTM-GWAS)方法,能够较全面地解析东北大豆种质群体百粒重性状QTL及其复等位变异,分析结果不仅可用于个别基因挖掘,还可用于群体遗传以及作物育种的优化组合设计等方面的研究。RTM-GWAS方法通过构建SNPLDB标记来检测资源群体的复等位变异,从而提高检测功效。本研究检测的76个位点上共计存在328个复等位变异(表3),平均每个位点存在4.3个复等位变异。与以往GWAS基于的SNP标记仅有2种变异相比,SNPLDB标记的复等位性更符合资源群体遗传特性。其次,RTM-GWAS方法采用多位点模型检测全基因组QTL,相比以往单位点模型方法,不仅提高了检测功效,还将检测位点的表型变异解释率控制在性状遗传率范围内。本研究定位到与百粒重关联的SNPLDB位点中61个主效显著位点共解释65.40%的表型变异。而以往方法往往只能检测到个别位点,例如COPLEY等[34]利用67 594个SNP标记仅定位到了5个百粒重相关位点。另外,本研究有34个SNPLDB标记位点与30个已报道QTL重叠,其余42个为本研究新检测位点。

表4 百粒重性状相关大效应QTL和候选基因
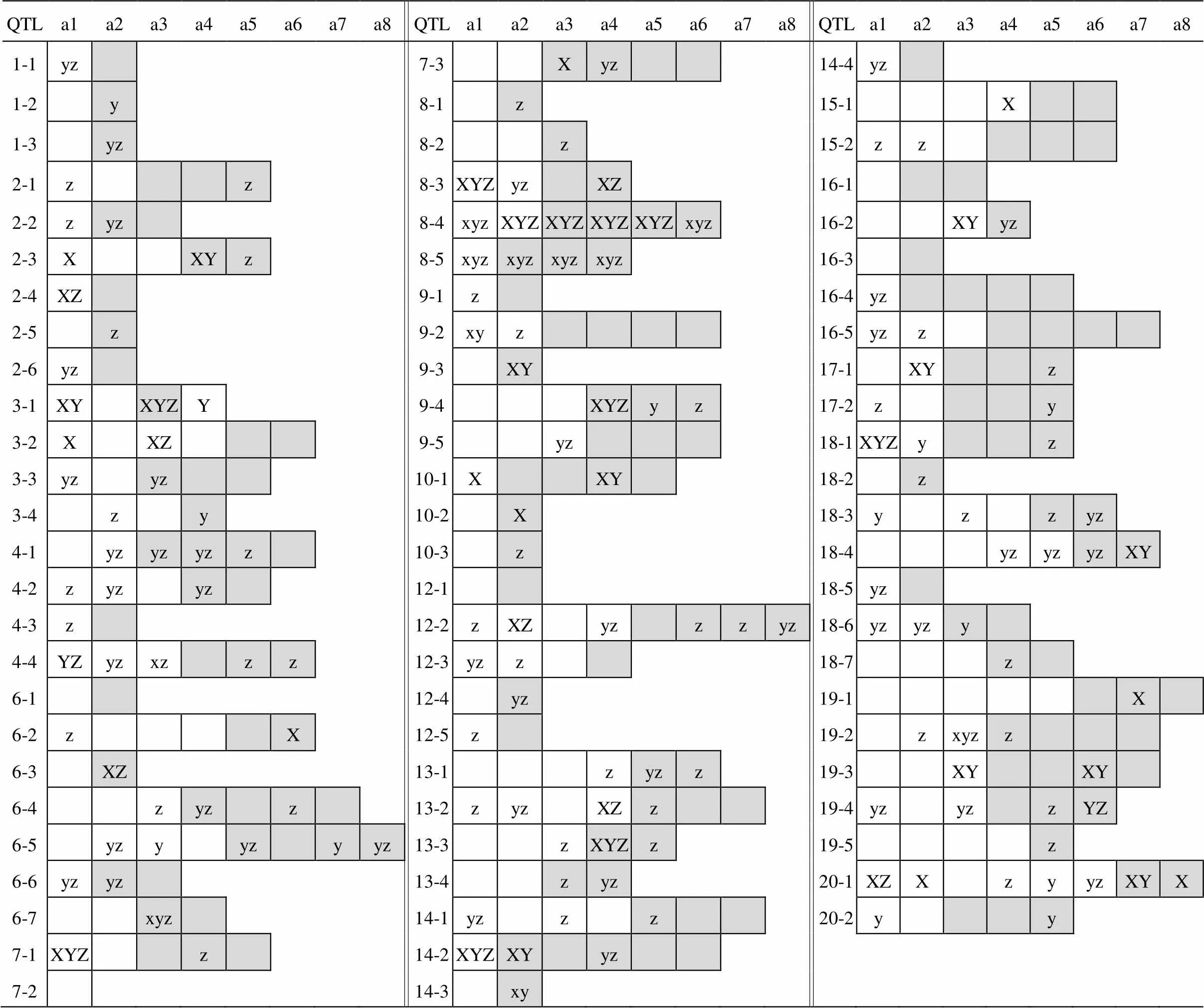
表5 百粒重QTL-等位变异在熟期组间的变化
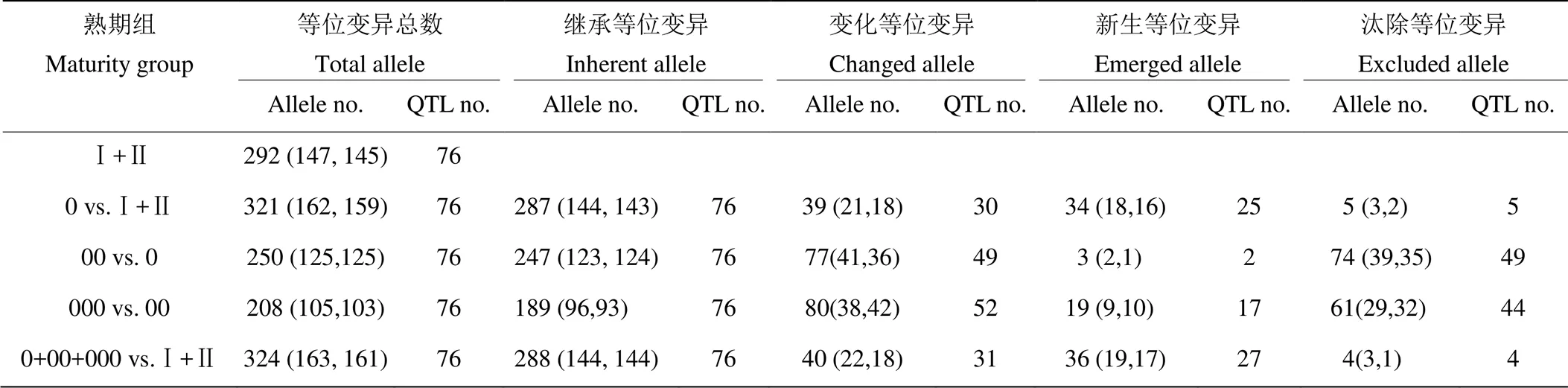
表格的上半部分:a1—a8表示每个QTL等位变异的编号,由a1至a8效应依次增大。表格中白色单元格表示负效应等位变异,灰色单元格表示正效应等位变异,没有大写字母的单元格表示MGI+Ⅱ的所有等位变异。带有小写字母“x”“y”“z”的单元格分别表示MG0、MG00、MG000 3个熟期组中汰除的等位变异(与MGI+Ⅱ相比)。带有大写字母“X”“Y”“Z”的单元格分别表示MG0、MG00、MG000 3个熟期组中新生的等位变异(与MGI+Ⅱ相比,且所有新生等位变异都不存在于MGI+Ⅱ中)。QTL名称列为简化后的名称,例如1-1,省去了“”。表格的下半部分:等位变异数目一列中,括号外面数字表示等位变异数目,括号内分别表示负效应和正效应的个数。继承等位变异个数表示由被比较的熟期组传递给各个待比较熟期组亚群的等位变异数目。变化的等位变异包括新生和汰除2种类型的等位变异
In the upper part: a1-a8 are the alleles of each QTL, arranged in a rising order according to their effect value. All the white cells represent alleles with negative effect and all the grey cells represent alleles with positive effect, and the cells without capital letters represent all the alleles of MGI+Ⅱ. The cells with lowercase “x” “y” “z” are alleles excluded in MG0, MG00, MG000 (vs. MGI+Ⅱ), respectively. The uppercase of “X” “Y” “Z” in cells means the alleles emerged in MG0, MG00, MG000 (vs. MGI+, but not exist in MGI+Ⅱ), respectively. In the column of QTL, the QTL name is simplified, such as 1-1, with “” omitted. In the lower part: In columns of Alleles, the number outside parentheses is the number of alleles, and the number in parentheses is the number of negative and positive alleles, respectively. Inherent allele means alleles passed from the compared MG. Changed allele includes the alleles excluded and emerged
候选基因分析共筛选到了137个与大豆百粒重相关的候选基因。这一数量远远超过前人研究结果。例如Wang等[35]利用SNP芯片定到了11个百粒重位点,但是只筛选到5个候选基因。Contreras-Sota等[36]仅得到了2个大豆百粒重相关的候选基因。Zhang等[37]总共筛选到6个候选基因。将前人的结果和RTM-GWAS方法的结果比较,充分证明了RTM-GWAS方法的优势和可行性。
本研究利用定位结果的QTL-等位变异体系对大豆各熟期组百粒重性状的遗传机制进行了研究,分析表明各熟期组间百粒重的变异幅度变化较大,主要是因为等位变异在新熟期组形成过程中发生了新生或汰除,比如,等位变异由熟期组00向熟期组000过度过程中第14染色体上的QTL的第1号等位变异(效应值为-7.39,是全部76个位点的328个等位变异中最大的负效等位变异,等位变异的顺序按照效应值从小到大排列)的汰除使得熟期组000中百粒重的最小值得到了大大提升,另外4个QTL,的第5号等位变异,的第6号等位变异,的第2号等位变异,的第5号等位变异(以上4个QTL的4个等位变异均为相应QTL位点上效应值最大的等位变异)的汰除也使得熟期组000百粒重的最大值由熟期组00的24.8 g降到21.8 g。从百粒重均值来看,各熟期组间变化不大,这主要是因为等位变异在传递过程中正效和负效等位变异几乎是同等数量的新生或汰除,但是比较发现各成熟期组中新生和汰除的等位变异不尽相同,发生了很大变化,说明百粒重的遗传机制在各熟期组间发生了变化。本研究为研究群体间等位变异的迁移汰除以及群体结构的变化提供了新思路。
4 结论
东北大豆种质群体中检测到76个大豆百粒重相关SNPLDB标记位点,共存在328个等位变异,其中61个主效显著位点解释了65.40%表型变异(大效应和小效应位点分别为18和43个,解释表型变异的52.15%和13.25%),68个与环境互作效应显著位点解释了17.46%的表型变异。所检测的34个SNPLDB标记位点与已报道30个QTL重叠。基于检测的SNPLDB标记位点,共注释到137个百粒重相关候选基因。各熟期组百粒重均值变化不大,但是QTL-等位变异比较分析显示各熟期组间百粒重的遗传结构发生了变化。
[1] 陈强, 闫龙, 冯燕, 邓莹莹, 侯文焕, 刘青, 刘兵强, 杨春燕, 张孟臣. 大豆百粒重QTL定位及多样性评价. 中国农业科学, 2016, 49(9): 1646-1656.
CHEN Q, YAN L, FENG Y, DENG Y Y, HOU W H, LIU Q, LIU B Q, YANG C Y, ZHANG M C. QTL Mapping and diversity evaluation of soybean 100-seed weight., 2016, 49(9): 1646-1656. (in Chinese)
[2] LU X, XIONG Q, CHENG T, LI Q T, LIU X L, BI Y D, LI W, ZHANG W K, MA B, LAI Y C, DU W G, MAN W Q, CHEN S Y, ZHANG J S. A PP2C-1 allele underlying a quantitative trait locus enhances soybean 100-seed weight., 2017, 10(5): 670-684.
[3] 汪霞, 李广军, 李河南, 艮文全, 章元明. 大豆百粒重QTL定位. 作物学报, 2010, 36(10): 1674-1682.
WANG X, LI G J, LI H N, GEN W Q, ZHANG Y M. QTL mapping for soybean 100-seed weight., 2010, 36(10): 1674-1682. (in Chinese)
[4] 齐照明, 孙亚男, 陈立君, 郭强, 刘春燕, 胡国华, 陈庆山. 基于Meta分析的大豆百粒重的QTLs定位. 中国农业科学, 2009, 42(11): 3795-3803.
QI Z M, SUN Y N, CHEN L J, GUO Q, LIU C Y, HU G H, CHEN Q S. Meta-analysis of 100-seed weight QTLs in soybean., 2009, 42(11): 3795-3803. (in Chinese)
[5] MIAN M A R, BAILEY M A, TAMULONIS J P, SHIPE E R, CARTER T E, PARROTT W A, ASHLEY D A, HUSSEY R S, BOERMA H R. Molecular markers associated with seed weight in two soybean populations., 1996, 93: 1011-1016.
[6] 傅蒙蒙, 王燕平, 任海祥, 王德亮, 包荣军, 杨兴勇, 田忠艳, 曹景举, 傅连舜, 程延喜, 苏江顺, 孙宾成, 杜维广, 赵团结, 盖钧镒. 东北春大豆熟期组的划分与地理分布. 大豆科学, 2016, 35(2): 181-192.
FU M M, WANG Y P, REN H X, WANG D L, BAO R J, YANG X Y, TIAN Z Y, CAO J J, FU L S, CHENG Y X, SU J S, SUN B C, DU W G, ZHAO T J, GAI J Y. A study on criterion, identification and distribution of maturity groups for spring-sowing soybeans in Northeast China., 2016, 35(2): 181-192. (in Chinese)
[7] MANSUR L M, ORF J H, CHASE K, JARVIK T, CREGAN P B, LARK K G. Genetic mapping of agronomic traits using recombi-nant inbred lines of soybean., 1996, 36: 1327-1336.
[8] CSANÁDI G, VOLLMANN J, STIFT G, LELLEY T. Seed quality QTLs identified in a molecular map of early maturing soybean., 2001, 103: 912-919.
[9] 宛煜嵩. 大豆遗传图谱的构建及若干农艺性状的 QTL 定位分析[D]. 北京: 中国农业科学院, 2002.
WAN Y S. Construction of soybean genetie map and QTL analysis of some agronomic traits[D]. Beijing: Chinese Academy of Agrieultural Sciences, 2002. (in Chinese)
[10] 孙亚男, 仕相林, 蒋洪蔚, 孙殿军, 辛大伟, 刘春燕, 胡国华, 陈庆山. 大豆百粒重QTL的上位效应和基因型×环境互作效应. 中国油料作物学报, 2012, 34(6): 598-603.
SUN Y N, SHI X L, JIANG H W, SUN D J, XIN D W, LIU C Y, HU G H, CHEN Q S. Epistatic effects and qE interaction effects of QTLs for 100-seed weight in soybean., 2012, 34(6): 598-603. (in Chinese)
[11] SUN Y N, PANN J B, SHI X L, DU X Y, LIU Q, QI Z M, JIANG H W, XIN D W, LIU C Y, HU G H, CHEN Q S. Multi-environment mapping and meta-analysis of 100-seed weight in soybase., 2012, 39(10): 9435-9443.
[12] KASTOORI R R, JEDLICKA J, GRAEF G L, WATERS B M. Identification of new QTLs for seed mineral, cysteine, and methionine concentrations in soybean [(L.) Merr.]., 2014, 34(2): 431-445.
[13] KATO S, SAYAMA T, FUJII K, YUMOTO S, KONO Y, HWANG T Y, KIKUCHI A, TAKADA Y, TANAKA Y, SHIRAIWA T, ISHIMOTO M. A major and stable QTL associated with seed weight in soybean across multiple environments and genetic backgrounds., 2014, 127(6):1365-1374.
[14] HAO D R, CHENG H, YIN Z T, CUI S Y, ZHANG D, WANG H, YU D Y. Identification of single nucleotide polymorphisms and haplotypes associated with yield and yield components in soybean () landraces across multiple environments.,2012, 124(3): 447-458.
[15] ZHOU Z, JIANG Y, WANG Z. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean., 2015, 33: 408.
[16] SONAH H, O’DONOUGHUE L, COBER E, RAJCAN I, BELZILE B. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soyabean., 2015, 13(2): 211-221.
[17] GUPTA P K, ROY J K, PRASAD M. Single nucleotide polymorphisms: a new paradigm for molecular marker technology and DNA polymorphism detection with emphasis on their use in plants., 2010, 80(4): 524-535.
[18] NACHMAN M W. Single nucleotide polymorphisms and recombination rate in humans., 2001, 17(9): 481-485.
[19] ZENG Z B. Precision mapping of quantitative trait loci., 1994, 136(4): 1457-1468.
[20] AUDIC S, CLAVERIE J M. The significance of digital gene expression profiles., 1997, 7(10): 986-995.
[21] BENJAMINI Y, DANIEL Y. The control of the false discovery rate in multiple testing under dependency., 2001, 4(29): 1165-1188.
[22] HE J B, MENG S, ZHAO T J, XING G N, YANG S P, LI Y, GUAN R Z, LU J J, WANG Y F, XIA Q J, YANG B, GAI J YAn innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding.,2017, 130(11): 2327-2343.
[23] ZHANG Y H, HE J B, WANG Y F, XING G N, ZHAO J M, LI Y, YANG S P, PALMER R G, ZHAO T J, GAI J Y. Establishment of a 100-seed weight quantitative trait locus–allele matrix of the germplasm population for optimal recombination design in soybean breeding programmes.,2015, 66(20): 6311-6325.
[24] LI S G, CAO Y C, HE J B, ZHAO T J, GAI J Y. Detecting the QTL‑allele system conferring flowering date in a nested association mapping population of soybean using a novel procedure., 2017, 130(11): 2297-2314.
[25] PAN L Y, HE J B, ZHAO T J, XING G N, WANG Y F, YU D Y, CHEN S Y, GAI J Y. The novel restricted two-stage multi-locus GWAS procedure efficient QTL detection of flowering date in a soybean RIL population using the novel restricted two‑stage multi‑locus GWAS procedure., 2018, 131(12): 2581-2599.
[26] 熊冬金. 中国大豆育成品种(1923-2005)基于系谱和SSR标记的遗传基础研究[D]. 南京: 南京农工业大学, 2009.
XIONG D J. Studies on the genetic bases of Chinese soybean cultivars released durling 1923-2005 based on pedigree and SSR marker analysis[D]. Nanjing: Nanjing Agricultural University, 2009. (in Chinese)
[27] ANDOLFATTO P, DAVISON D, EREZYILMAZ D, HU T T, MAST J, SUNAYAMA-MORITA T, STERN D L. Multiplexed shotgun genotyping for rapid and efficient genetic mapping., 2011, 21(4): 610-617.
[28] LI R, YU C, LI Y, LAM T W, YIU S M, KRISTIANSEN K, WANG J. SOAP2: an improved ultrafast tool for short read alignment., 2009, 25(15): 1966-1967.
[29] JEREMY S, STEVEN B. C, JESSICA S, JIAN X M, THERESE M, WILLIAM N, DAVID L. H, QI J S, JAY J. T, JIANLIN C, DONG X, UFFE H, GREGORY D. M, YEISOO Y, TETSUYA S, TAISHI U, MADAN K. B, DEVINDER S, BABU V, ERIKA L, MYRON P, DAVID G, SHU S Q, DAVID G, KERRIE B, MONTONA F, BRIAN A, DU J C, TIAN Z X, ZHU L C, NAVDEEP G, TRUPTI J, MARC L, ANAND S, ZHANG X C, KAZUO S, HENRY T. N, ROD A. W, PERRY C, JAMES S, JANE G, DAN R, GARY S, RANDY C. S, SCOTT A. JGenome sequence of the palaeopolyploid soybean., 2010, 463(14): 178-183.
[30] YI X, LIANG Y, HUERTA-SANCHEZ E, JIN X, CUO Z X, POOL J E, XU X, JIANG H, VINCKENBOSCH N, KORNELIUSSEN T S, ZHENG H, LIU T, HE W, LI K, LUO R, NIE X, WU H, ZHAO M, CAO H, ZOU J, SHAN Y, LI S, YANG Q, ASAN, NI P, TIAN G, XU J, LIU X, JIANG T, WU R, ZHOU G, TANG M, QIN J, WANG T, FENG S, LI G, HUASANG, LUOSANG J, WANG W, CHEN F, WANG Y, ZHENG X, LI Z, BIANBA Z, YANG G, WANG X, TANG S, GAO G, CHEN Y, LUO Z, GUSANG L, CAO Z, ZHANG Q, OUYANG W, REN X, LIANG H, ZHENG H, HUANG Y, LI J, BOLUND L, KRISTIANSEN K, LI Y, ZHANG Y, ZHANG X, LI R, LI S, YANG H, NIELSEN R, WANG J. Sequencing of 50 human exomes reveals adaptation to high altitude., 2010, 329(5987): 75-78.
[31] SCHEET P, STEPHENS M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase., 2006, 78(4): 629-644.
[32] KUMAR S, DUDLEY J, NEI M, TAMURA K. MEGA: A biologist- centric software for evolutionary analysis of DNA and protein sequences., 2008, 9: 299-306.
[33] 盖钧镒, 汪越胜, 张孟臣, 王继安, 常汝镇. 中国大豆品种熟期组划分的研究. 作物学报, 2001, 27(3): 286-292.
GAI J Y, WANG Y S, ZHANG M C, WANG J A, CHANG R Z. Studies on the classification of maturity groups of soybean in China., 2001, 27(3): 286-292. (in Chinese)
[34] COPLEY T R, DUCEPPE M O, O’DONOUGHUE L S. Identification of novel loci associated with maturity and yield traits in early maturity soybean plant introduction lines., 2018, 19(1): 167.
[35] WANG J, CHU S, ZHANG H, ZHU Y, CHENG H, YU D Y. Development and application of a novel genome-wide SNP array reveals domestication history in soybean., 2016, 6: 20728.
[36] CONTRERAS-SOTO R I, MORA F, OLIVEIRA M A R, HIGASHI W, SCAPIM C A, SCHUSTER I. A genome-wide association study for agronomic traits in soybean using SNP markers and SNP-based haplotype analysis., 2017, 12(2): e0171105.
[37] ZHANG J, SONG Q, CREGAN P B, JIANG J L. Genome-wide association study, genomic prediction and marker-assisted selection for seed weight in soybean ()., 2016, 129: 117.
Genome-wide QTL-allele Dissection of 100-seed weight in the Northeast China Soybean Germplasm Population
HAO XiaoShuai1, FU MengMeng1, LIU ZaiDong1, HE JianBo1, WANG YanPing2, REN HaiXiang2, WANG DeLiang3, YANG XingYong4, CHENG YanXi5, DU WeiGuang2, GAI JunYi1
(1Soybean Research Institute, Nanjing Agricultural University/National Center for Soybean Improvement/Key Laboratory of Biology and Genetic Improvement of Soybean (General), Ministry of Agriculture/State Key Laboratory for Crop Genetics and Germplasm Enhancement/Jiangsu Collaborative Innovation Center for Modern Crop Production, Nanjing 210095;2Mudanjiang Branch of Heilongjiang Academy of Agricultural Sciences/Mudanjiang Experiment Station of the National Center for Soybean Improvement, Mudanjiang 157041, Heilongjiang;3Heilongjiang Academy of Land-reclamation Sciences, Jiamusi 154007, Heilongjiang;4Keshan Branch of Heilongjiang Academy of Agricultural Sciences, Keshan 161606, Heilongjiang;5Changchun Academy of Agricultural Sciences, Changchun 130111)
【】A genome-wide association study in the Northeast China soybean germplasm population was conducted for a relatively thorough detection of the QTL-allele constitution of 100-seed weight, which may provide a theoretical basis for soybean breeding for seed size improvement.【】In the present study, a total of 290 soybean accessions that were frequently used for soybean breeding and production in the Northeast China were tested in 2013 and 2014 for 100-seed weight at four locations, including Keshan, Mudanjiang, Jiamusi and Changchun, which are all in the second sub-ecoregion of the Northeast China. RAD-seq (restriction site-associated DNA sequencing) was used for SNP genotyping, and 82 966 high-quality SNPs were obtained after filtering and imputation. According to the RTM-GWAS (restricted two-stage multi-locus genome-wide association analysis) method, firstly a total of 15 546 multi-allelic SNPLDBs were constructed, and then a multi-locus model was used for genome-wide association study of 100-seed weight. The genes near (within 50kb) the detected SNPLDBs were analyzed, and candidate genes for 100-seed weight were identified and annotated according to Chi-square test of independence between the SNPs within genes and the detected SNPLDBs. Finally, genetic differentiation among maturity groups were investigated based on the detected QTL-allele system of 100-seed weight.【】The 100-seed weight of the present population ranged from 18.3 to 20.7 g, and the trait heritability was 92.3%. A total of 76 SNPLDBs were detected to be associated with 100-seed weight, among which there were 15 SNPLDBs with non-significant main effect and the 61 SNPLDBs with significant main effect explained 65.40% phenotypic variation. There were 68 SNPLDBs that had significant interaction effect with environment and explained 17.46% phenotypic variation. In addition, 34 out of 76 detected SNPLDBs overlapped 30 previously reported QTLs and 42 SNPLDBs were novel loci. A total of 137 candidate genes for 100-seed weight were annotated in the detected SNPLDB regions, and functional annotation showed that these genes were not only involved in regulation of 100-seed weight, but also involved in primary metabolism, translation, protein modification, material transport, stress response and signal transduction, etc. Although there was no obvious difference in the 100-seed weight among different maturity groups, genetic differentiation analysis showed varying changes of allele emergence and exclusion in QTL-allele structure of 100-seed weight among maturity groups. 【】The RTM-GWAS method used in the present study provided a relatively thorough detection of genome-wide QTLs and their multiple alleles for 100-seed weight in the Northeast China soybean germplasm population. The 100-seed weight of the Northeast China soybean germplasm population was controlled by a large number of QTLs with large significant interaction effect with environment, and there was also abundant multiple allelic variation in these QTLs. The QTL-allele matrix established from RTM-GWAS provided an efficient tool for population genetics and evolution study.
soybean [(L.) Merr.]; 100-seed weight; RTM-GWAS; QTL-allele matrix; candidate gene

10.3864/j.issn.0578-1752.2020.09.003
2019-09-09;
2020-01-02
国家自然科学基金(31701447)、国家作物育种重点研发计划(2017YFD0101500,2017YFD0102002)、长江学者和创新团队发展计划(PCSIRT_17R55)、教育部111项目(B08025)、中央高校基本科研业务费项目(KYT201801)、农业部国家大豆产业技术体系CARS-04、江苏省优势学科建设工程专项、江苏省JCIC-MCP项目
郝晓帅,E-mail:15850563928@163.com。通信作者贺建波,E-mail:hjbxyz@gmail.com。通信作者盖钧镒,E-mail:sri@njau.edu.cn
(责任编辑 李莉)
猜你喜欢
军事文摘(2022年16期)2022-08-24
作物学报(2022年6期)2022-04-08
国际医学放射学杂志(2021年5期)2021-10-22
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
支部建设(2020年15期)2020-07-08
第一财经(2017年36期)2017-09-25
百科知识(2015年18期)2015-09-10
小星星·阅读100分(高年级)(2015年4期)2015-05-26
