
赖氨酸特异性去甲基酶1反苯环丙胺类不可逆抑制剂的研究进展
2020-05-29 04:26:14高琦冰刘超亿孙旭东马超亚王志正郑甲信丁丽娜
药学进展 2020年4期
高琦冰,刘超亿,孙旭东,马超亚,王志正,郑甲信,丁丽娜
(郑州大学药学院 药物关键制备技术教育部重点实验室,河南 郑州 450001)
组蛋白修饰是指组蛋白在相关酶作用下发生甲基化、乙酰化、磷酸化、腺苷酸化、泛素化等修饰的过程。一直以来,组蛋白修饰中的大多数修饰都被认为是可逆过程,但甲基化修饰却是一个不可逆过程。直到2004年,随着组蛋白赖氨酸特异性去甲基酶1(lysine specific demthylase1,LSD1)的发现,甲基化修饰的可逆性才被公布于世[1]。LSD1是一种黄素腺嘌呤二核苷酸(flavin adenine dinucleotide,FAD)依赖性胺氧酶,LSD1对甲基化状态的调控会影响相关基因的表达,从而调节相应的生理学过程——LSD1能特异性催化去除组蛋白H3K4和H3K9的单、双甲基,调节组蛋白和其他蛋白间的相互作用,进而影响基因转录的激活、抑制与染色体失活等过程,被称为细胞深处的基因“开关”。另外,LSD1还能去除P53、DNA 甲基转移酶、E2F转录因子1、信号转导和转录激活因子3等蛋白的甲基,进而调节下游细胞的生理功能[2-5]。研究显示,LSD1在多种肿瘤细胞中高表达[6-7],如胃癌、肺癌、前列腺癌、神经细胞瘤、乳腺癌、膀胱癌等,因此开发有效的LSD1抑制剂已成为肿瘤预防和治疗的新方向。目前已有多种LSD1抑制剂被报道,根据其作用方式的不同可分为两大类,一类是与LSD1中辅酶FAD共价结合来发挥抑制作用的不可逆抑制剂,另一类是非共价地结合在LSD1的底物结合位点或FAD结合位点来发挥抑制作用的可逆抑制剂。不可逆抑制剂较可逆抑制剂结合更稳定、特异性高且化合物不易脱靶,因此不可逆抑制剂成为LSD1抑制剂研究的更优方向。目前已有数个LSD1不可逆抑制剂进入临床试验,进一步证实了LSD1不可逆抑制剂的研究价值与成药性。
LSD1与单胺氧化酶(monoamine oxidase,MAO)在催化区域具有45%的相同序列,在序列结构上具有高度同源性[8],因此针对二者催化区域以及结构上的相似性,研究人员开始研究MAO抑制剂对LSD1的抑制作用。2006年Lee 等[9]报道了MAO抑制剂对LSD1的抑制活性测试结果,发现反苯环丙胺类(tranylcypromine,TCP)不可逆MAO抑制剂对LSD1的抑制效果最好。但相比对MAO的抑制效果,TCP 对LSD1 的活性和选择性均很差,因此众多课题组纷纷投入到对TCP的结构改造工作中,试图得到LSD1的特异性强效抑制剂。各课题组从不同层面对此类抑制剂展开探索,主要包括:1)TCP 类化合物的抑制机制以及与FAD 结合模式;2)TCP 类抑制剂的设计优化。本文将针对上述两方面逐一概述。
1TCP类化合物的抑制机制以及与FAD 结合模式
2007年Schmidt 等[10]报道了TCP对LSD1的抑制机制,经实验以及对TCP-FAD复合物的质谱分析证实了TCP对LSD1的不可逆抑制作用是通过与FAD共价结合实现的。推测其反应机制为:TCP中环丙烷经单电子转移开环进而共价结合至FAD-C4位,生成亚胺中间体,然后经水解得到TCP-FAD复合物。由于TCP中环丙烷结构的不对称性,导致开环位置也相应不同,故存在2种C4位加成机制(路径1、2,见图1)。
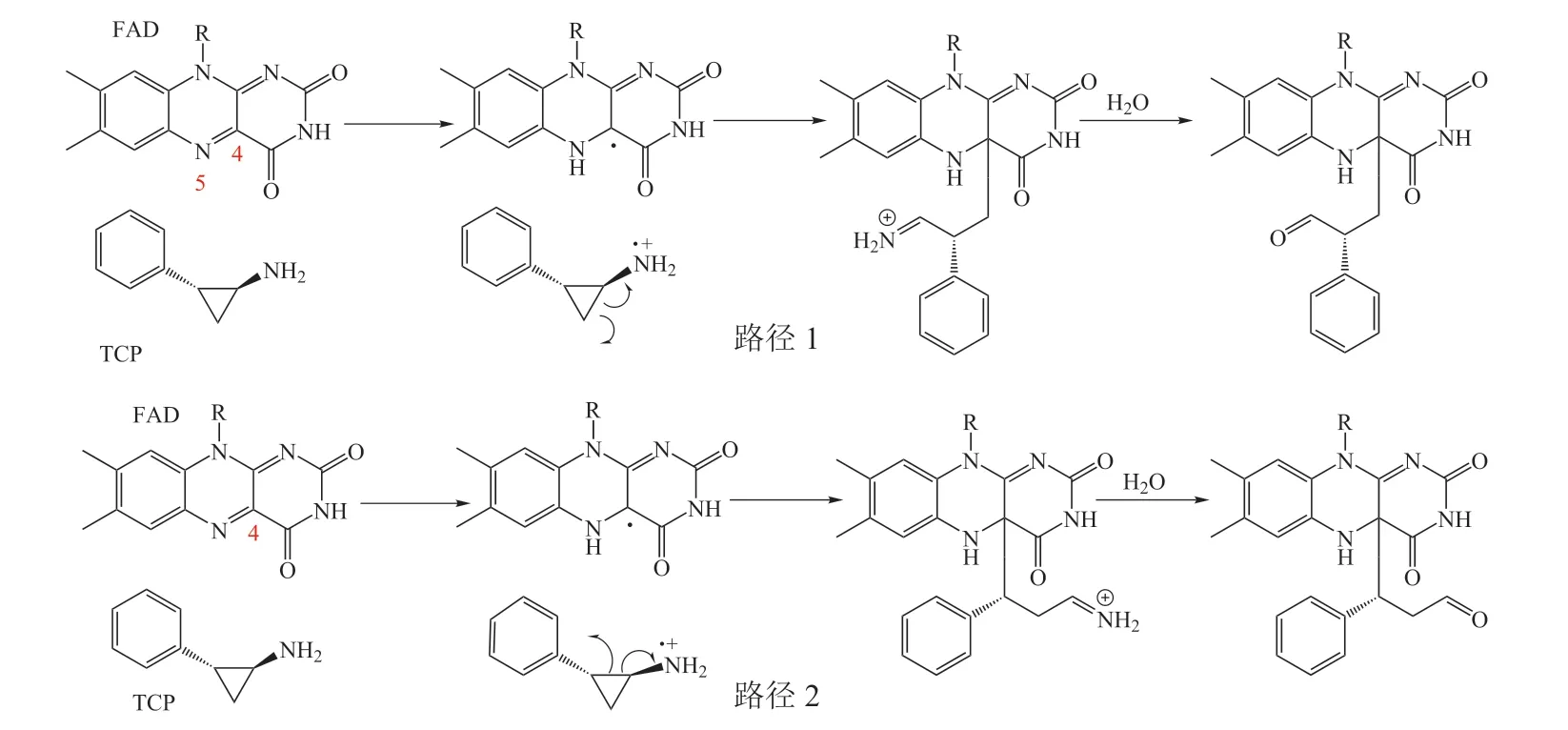
图1 TCP 与FAD-C4 位的2种加成机制[9]Figure 1 Two addition mechanisms of TCP and FAD-C4 site[9]
同年,Yang 等[11]解析得到TCP-FAD晶体,分析确证了TCP连接至FAD-C4位复合物的存在,但电子密度图显示FAD的N5、C4原子位置均发生了结构修饰,即这2个原子均与TCP成键。由此推测TCP-FAD的结晶中还存在TCP 共价结合至FAD的N5、C4位原子的五元环结构,随后结合质谱、紫外可见光谱的分析结果证实了五元环晶体模型的存在,且为主要存在形式。推测五元环结构形成机制为:同样先经由单电子转移机制致使环丙烷开环,生成苄基自由基与亚胺中间体,随后经自由基反应结合至FAD,同时亚胺水解进而与FAD成环形成TCPFAD五元环结构(见图2)。
随后Mimasu 等[12]在提高结晶解析度的条件下得到更精细的TCP与LSD1的复合晶体结构。然而经过分析发现TCP与LSD1中FAD结构的五元环结合模式、C4结合模式均无法完全匹配Fo-Fc 电子密度图,鉴于此,他们推测结晶中可能还同时存在TCP 仅进攻FAD-N5 位的结合模式。推测其机制为:TCP 在环丙烷开环同时对FAD亲核加成,之后经氢转移、亚胺水解得到加成产物(见图3)。但吸收光谱显示五元环结构仍是TCP-FAD复合物的主要存在形式。
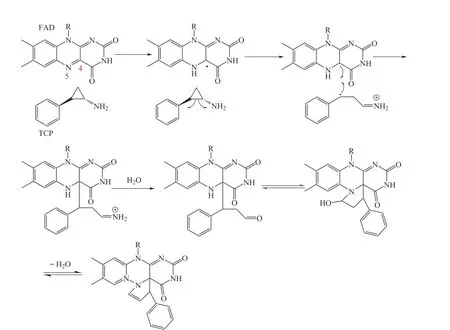
图2 TCP-FAD五元环结构形成机制[11]Figure2 Formation of five-membered ring structure of TCP-FAD[11]
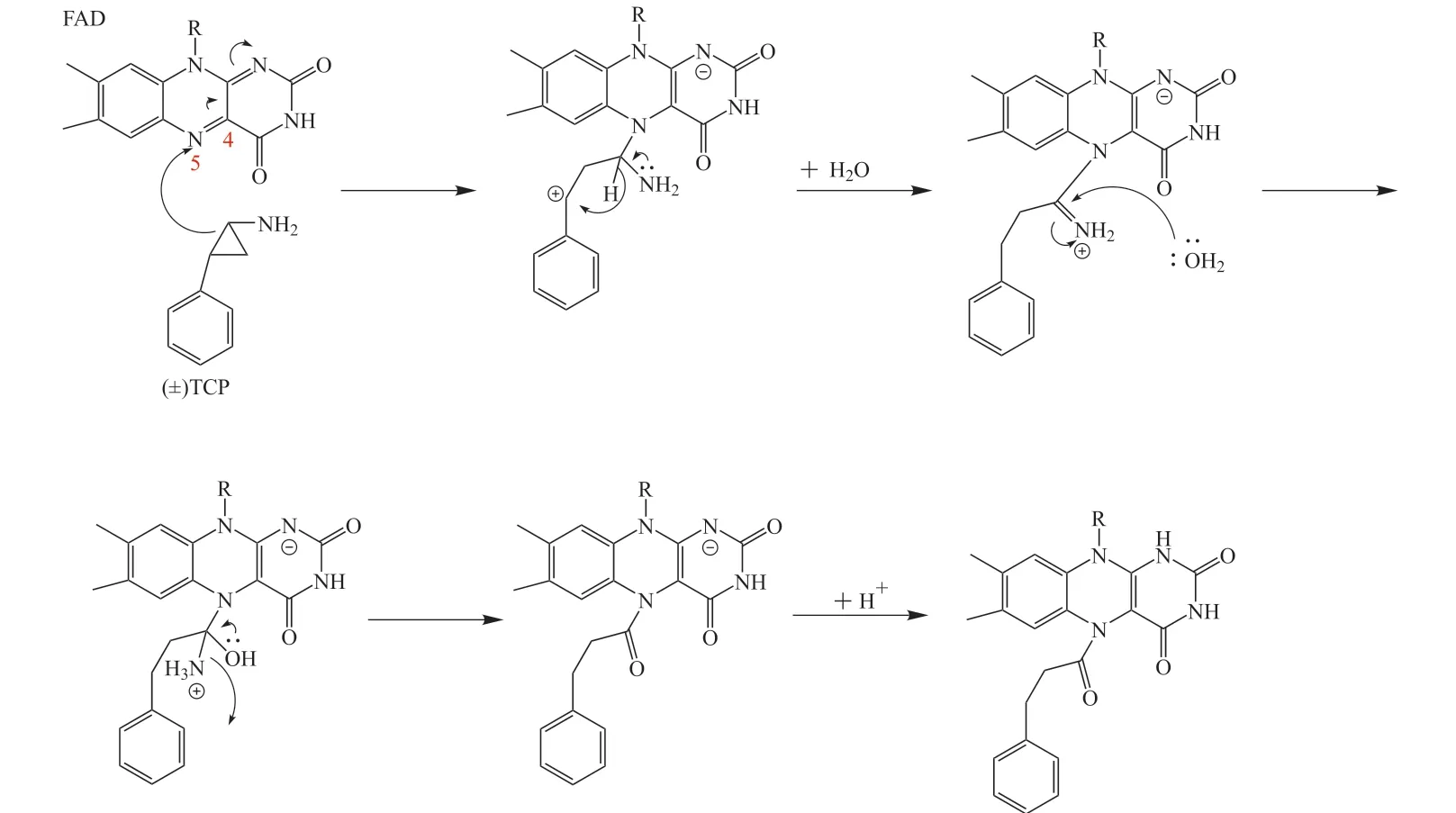
图3 TCP仅进攻FAD-N5位的结合模式的机制[12]Figure3 Binding mode of TCP attacking the FAD-N5 site only[12]
TCP-FAD的3种结合模式在2014年Vianello等[13]改造得到的环丙胺α位取代的TCP衍生物与LSD1的复合物晶体结构中均被证实。但环丙胺α位被取代后无法经单电子转移机制生成亚胺中间体,所以该课题组又提出此类抑制剂经自由基机制直接开环而共价结合至FAD的方案(见图4)。
截至目前,TCP类抑制剂与LSD1辅酶FAD的3种结合模式在晶体结构中均已得到证实,但二者的结合机制尚无定论。晶体结构的解析在抑制剂特异性和活性的优化方面起到重要指导作用,在研究者尚无法得到其所设计合成的化合物与LSD1复合物晶体的情况下,通过分子模拟手段指导化合物进行改造就显现出优势。但化合物与LSD1结合机制的不确定性会给分子模拟带来很大的挑战,因此仍需对TCP类抑制剂的结合机制进行深入研究。
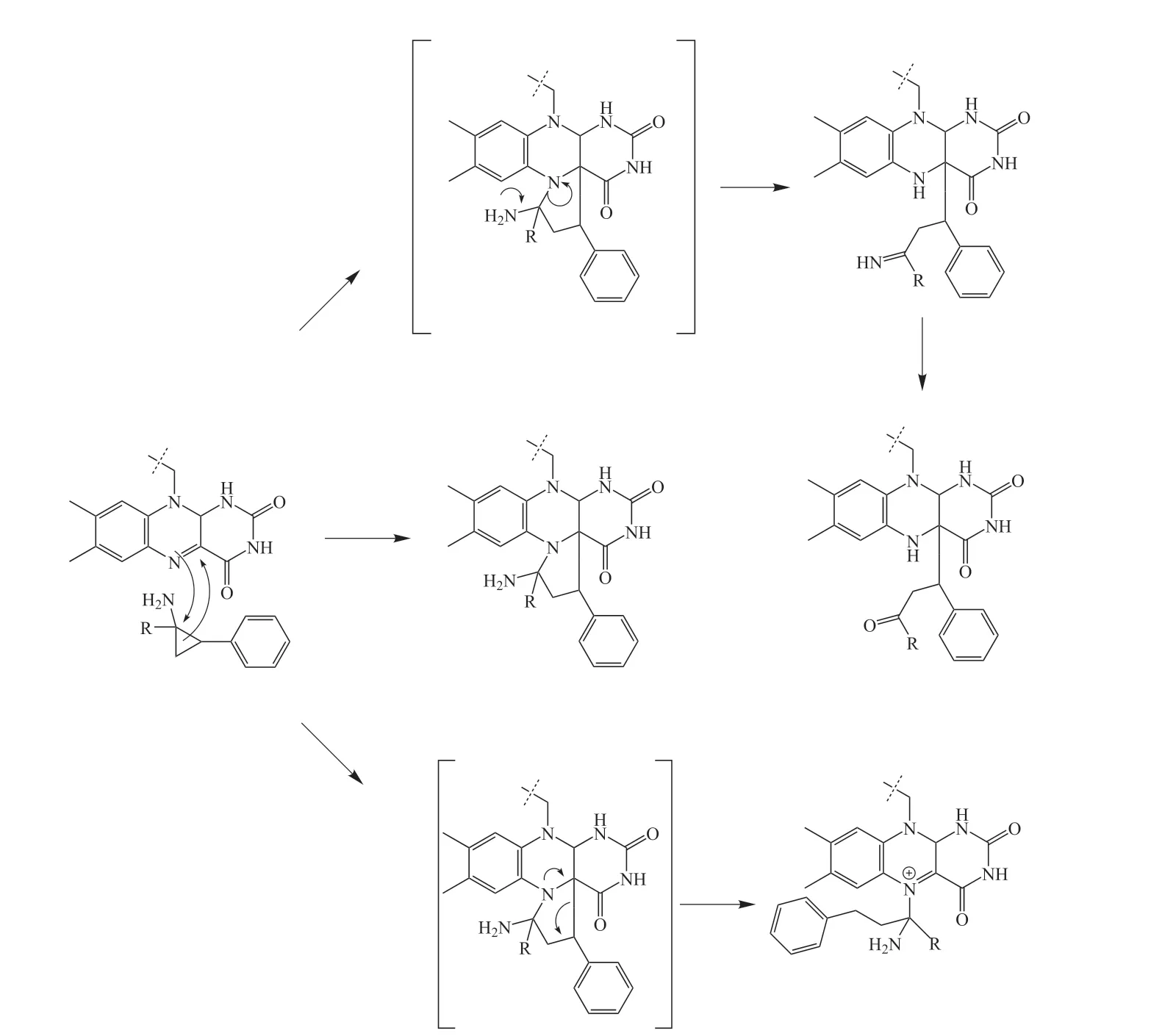
图4 环丙胺α位取代的TCP衍生物结合LSD1的自由基机制[13]Figure 4 Free radical mechanism of cyclopropyla mine α-substituted TCP derivative binding to LSD1[13]
2TCP类抑制剂的设计优化
TCP对MAO和LSD1的抑制作用均通过与其辅酶FAD共价结合而实现,但与FAD的结合方式却不同。对比2种酶与TCP的复合物晶体结构可以看出[11],TCP在LSD1中同时结合至FAD-C4/N5位,并以这种五元环方式为主要结合模式(PDB ID : 4UVB,见 图5);而TCP在MAO的B亚 型中只能结合至FAD-C4位(PDBID : 2XFU,见图6)。若TCP采取五元环的模式结合至MAO-B的FAD-C4/N5位,TCP的苯环将与Tyr435残基存在严重的立体碰撞(见图7),而结合至FAD-C4位不仅可以避免这种不利碰撞,同时苯环与周围的疏水残基Tyr435、Tyr398、Gln206、Tyr60、Phe343 等形成的疏水作用也有利于复合物保持稳定。
由于TCP类抑制剂是由LSD1的同源蛋白抑制剂发展而来,对其结构改造和优化主要集中在对LSD1的特异性和作用强度方面。如前所述,TCP类抑制剂在MAO和LSD1中采取了不同的结合方式,这一差异也成为TCP类抑制剂特异性改造方面的一个重要突破口,可基于2种酶活性位点的结构差异,通过在苯环上引入较大基团等方式以增强对LSD1的特异性。另外TCP-LSD1类复合物晶体结构表明TCP苯环位于由Val333、His564、Thr335、Thr810、Ala809、Tyr761等残基形成的较大疏水性空腔中(见图5),抑制剂上的苯环仅与Thr335和Thr810上的甲基存在很弱的范德华作用,且与周围残基没有形成广泛相互作用,这提示在苯环上引入疏水取代基,可能通过增强与周围残基的疏水作用从而提升抑制剂活性。
2.1TCP 苯环的结构修饰
2.1.1 基于活性位点空间结构差异进行的结构修饰基于LSD1和MAO活性位点的空间结构差异,在TCP 苯环上引入较大基团,可以增加与MAO活性位点残基的立体碰撞,从而增强对LSD1 的特异性。
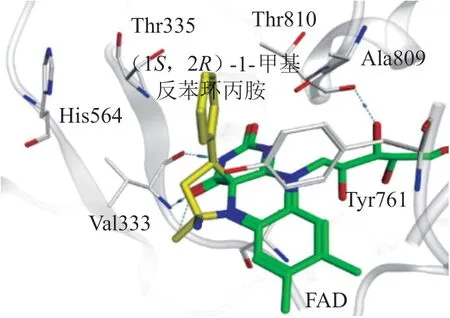
图5 (1S, 2R)-1-甲基反苯环丙胺与LSD1的FAD-C4/N5位结合模式图Figure 5 Binding mode of (1S, 2R)-1-methyl-tranylcypromine in the FAD-C4/N5sites of LSD1
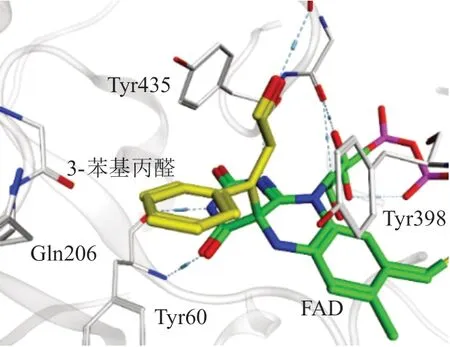
图6 3-苯基丙醛与MAO-B的FAD-C4 位结合模式图Figure 6 Binding mode of 3-phenylpropanal in FAD-C4 site of MAO-B
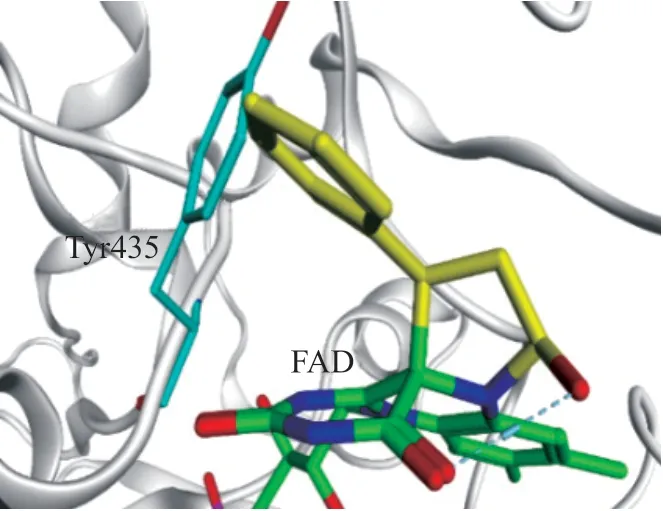
图7 TCP 与MAO-B的FAD-C4/N5位结合模式图Figure 7Binding mode of TCP inFAD-C4/N5sites of MAO-B
考虑到TCP 中环丙胺β位苯环的邻位取代对选择性的影响,2010 年Mimasu 等[14]设计合成得到邻位体积较大的苄氧基取代的化合物S1201(1),其对LSD1的活性和选择性(IC50=8.1μmol · L-1)相比TCP均有提高,晶体结构(PDBID : 3ABU)显示活性提高是由于引入的邻位苄氧基与Val333、Phe538、Thr335、His564等疏水残基的疏水作用稳定了复合物构象(见图8),选择性提高则是MAO活性位点残基与化合物1立体碰撞的结果。在晶体结构指导下,向化合物1苯环间位引入疏水F原子得到化合物S2101(2),其因与周围残基的疏水作用进一步增强,活性和选择性更好(IC50=0.99μmol · L-1)。
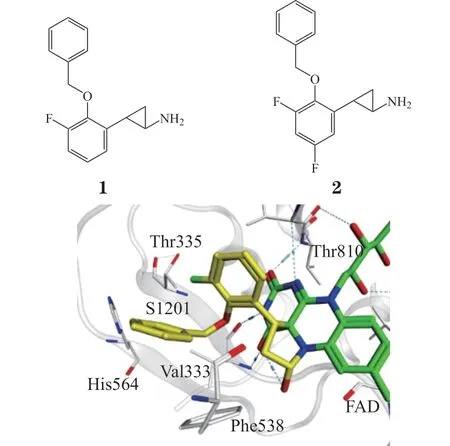
图8 化合物1-LSD1复合物构象图Figure 8 Complex conformation of compound 1-LSD1
同时,Binda 等[15]也采取在TCP苯环上引入不同性质的大体积取代基的方法以期增强对LSD1的选择性和活性,经过筛选获得2个活性最好的化合物3(IC50=0.0192μmol · L-1)和4(IC50=0.0227 μmol · L-1),相对于TCP活性明显提高。化合物4中由于支链的存在,导致其对活性位点狭窄的MAO-B无抑制活性,但同时作者推测由于缺乏氢键等特异性相互作用而使化合物4 对LSD1和MAO-A缺乏选择性。
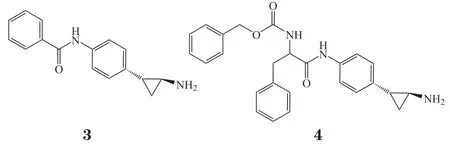
随后Valente[16]和Vianello[17]课题组分别在化合物3(反式结构)的基础上进行了结构改造。2015年Valente 等[16]合成了化合物3的异构体,经活性测试发现顺式化合物5、反式化合物6均有较强的LSD1抑 制活性(IC50分别为0.021和0.022μmol ·L-1),而且能诱导白血病NB4细胞中独立生长因子1B(growth factor independence-1B,gfi1b)和整合素αM(including integrin alpha M,itgam)基因的高表达,强烈抑制小鼠急性早幼粒细胞性白血病(acute promyelocytic leukemia,APL)细胞的克隆形成能力。
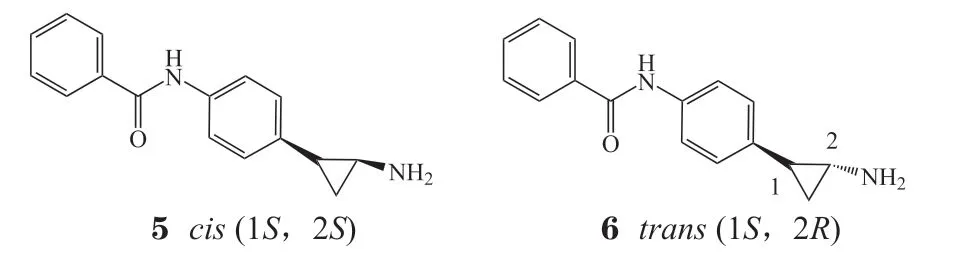
Vianello等[17]对化合物3的结构修饰主要是在与羰基相连的苯环上引入各种环状体系,苯环上的取代基可以在不改变化合物类药性的前提下,充分占据活性位点,进而提高化合物选择性和活性。最终得到的外消旋体7(IC50=0.1885μmol · L-1)虽然抑制活性弱于化合物3,但经小鼠体内试验及小鼠APL 模型中生物、细胞活性测试和初步药动学研究证实,其具有较高的LSD1选择性和良好的口服生物利用度,当浓度为250nmol · L-1时,其对APL细胞的抑制率为85%,浓度为500nmol · L-1时对人类白血病单核细胞系的抑制率为60%。随后合成得到化合物7的光学单体8(1S,2R),相比化合物7,其对LSD1 的抑制活性(IC50=0.084μmol · L-1)增强,而其他生物测试结果均与化合物7类似。化合物8的各项生物评价数据都为其进入临床前研究提供了一定依据。

2.1.2 基于晶体叠合进行的结构修饰2009年,Ueda等[18]将TCP-LSD1和N-炔丙基赖氨酸肽-LSD1晶体[19]进行了叠合,叠合显示TCP苯环与赖氨酸N原子重合(见图9),所以考虑用TCP代替N-炔丙基赖氨酸肽结构中与之重合的部分,并以醚键将二者相连,同时在赖氨酸N端引入疏水侧链以增强对LSD1的选择性。从而得到2个具有细胞活性的LSD1选择性抑制剂——化合物9(IC50=2.5μmol ·L-1)和10(IC50=1.9μmol · L-1)。

图9 TCP-LSD1和N-炔丙基赖氨酸肽-LSD1晶体叠合图Figure 9Superimposed image of TCP-LSD1and N-pro- pargyllysinepeptide-LSD1crystals
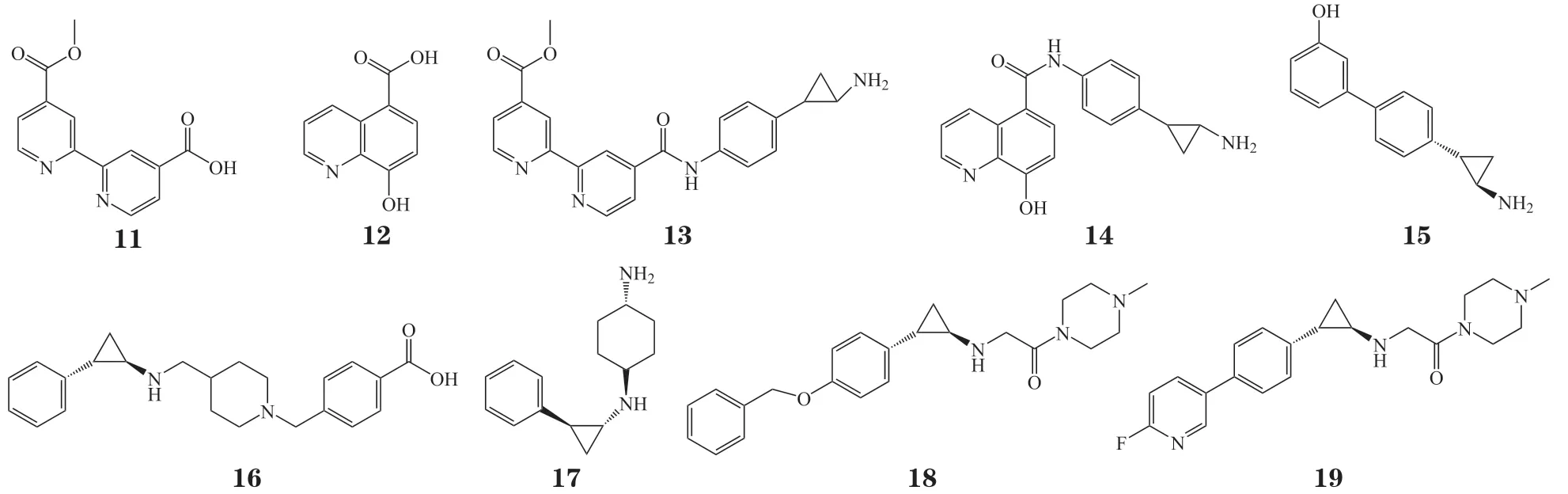
2.1.3 基于分子拼接原理进行的结构修饰目前有两类去甲基酶被报道,分别是LSD家族和含JmjC结构域的去甲基化酶家族。在前列腺癌中LSD1和JmjC 的JMJD2亚型与雄激素受体共表达、共定位。但单独JMJD2抑制剂不能抑制前列腺癌和结肠癌细胞的生长,当其与LSD1抑制剂NCL-2联合使用时则显示出抗增殖作用,表明LSD和JmjC抑制具有协同作用,因此基于分子拼接原理对LSD和JmjC抑制剂进行结构修饰,有望获得对二者均有抑制作用的高效抑制剂。2014年,Rotili 等[20]利用分子拼接原理将LSD1抑制剂TCP与2个竞争JmjC酶辅因子α-酮戊二酸的抑制剂4-羧基-4'-甲酯基-2,2'-联吡啶(11)[21]、5-羧基-8-羟基喹啉(12)[22]拼接得到2个泛-去甲基酶抑制剂13和14。活性测试结果显示,这2个化合物对LSD1和JmjC酶均有较好的抑制活性和特异性:化合物13和14 对LSD1的IC50均低于1μmol · L-1,对JmjC的JMJD2亚 型的IC50分别为2.7和1.2μmol · L-1,对其 他亚型的IC50则分别为8.5~76和3.9~31μmol · L-1。分子模拟推测此类化合物在LSD1蛋白和JmjC家族蛋白中发挥抑制作用的机制不同,TCP通过与FAD的共价结合来抑制LSD1活性,TCP对JmjC的抑制作用则通过喹啉或吡啶部分可逆地结合至其活性位点而实现。
2.1.4 基于筛选实验进行的结构修饰2013年,Liang等[23]以纯化的LSD1蛋白经体内LSD1去甲基化实验筛选得到高活性的TCP类抑制剂OG-L002(15,IC50=0.02μmol · L-1)。研究报道[24-25]LSD1作为一种转录调控复合物的重要组分,能激活单纯疱疹病毒(herpes simplex virus,HSV)、人类免疫缺陷病毒(human immunodeficiency virus,HIV)的基因表达,随后经细胞实验证实化合物15对LSD1的活性抑制确实能强烈抑制HSV 即刻早期基因(immediate early genes,IEGs)表达和体内病毒裂解性感染,即LSD1的活性抑制致使HSV、HIV 表达受阻,因此LSD1不仅是一种抗肿瘤靶标,同时可作为一种新型抗病毒靶标。
2.2TCP氨基的结构修饰
结合上述已报道的抑制剂和相关文章可以发现,对于TCP的结构修饰主要集中于苯环,而目前进入临床研究且已知结构的2个TCP类抑制剂均为氨基被修饰的化合物[25-26]。TCP氨基的结构修饰,通过在氨基位置引入取代基合成新的TCP衍生物,获得了高选择性和高抑制活性的抑制剂,但是合成方法存在一定难度且收率较低。
2007年,葛兰素史克公司经高通量筛选、结构优化得到具有高度选择性和抗肿瘤活性的化合物GSK2879552(16)[27-29],目前作为LSD1不可逆抑制剂进入临床试验,其口服用于治疗复发或难治性小细胞肺癌(small cell lung cancer,SCLC)和骨髓增生异常综合征(myelodysplastic syndromes,MDS)的研究分别处于Ⅰ期和Ⅱ期临床阶段[30]。
Oryzon 公司开发的化合物ORY-1001(17)[31-33]于2013年获得欧洲药品管理局孤儿药资格,其对LSD1具有高选择性和抑制活性,是目前已进入临床试验的LSD1不可逆抑制剂,并于2014年被罗氏公司收购获得其研发及商业化权利。目前其口服用于治疗急性髓系白血病的研究处于Ⅱ期临床阶段。
2012年Neelamegam 等[34]设计合成了一系列取代氨基的TCP衍生物,经多种实验测试确定了活性最高、选择性最好的2个化合物RN-1(18,IC50=0.01μmol · L-1)、RN-7(19,IC50=0.003μmol · L-1)。同时作者选用抑制剂18考察LSD1对长期记忆形成的影响,经实验证实RN系列化合物具有良好的血脑屏障渗透作用,会破坏长期记忆的形成,但不影响短期记忆,说明LSD1可能对长期记忆的形成起到正调节作用。
2.3TCP环丙基的结构修饰
目前对TCP苯环、氨基的结构改造已有相对深入的研究,对环丙烷部分的修饰却鲜有报道。2014年Vianello等[13]合成得到了一系列环丙胺α位亲水、疏水基取代的TCP 衍生物,经测试得到活性最好的化合物20(IC50=0.161μmol · L-1)。构效关系表明:疏水基团更有利于活性提高,化合物取代基体积适当增大可以有效提高LSD1的抑制活性和特异性。TCP环丙烷上取代基的变化影响LSD1抑制活性及特异性的这一现象为衍生物的结构修饰提供了新思路。
在TCP与LSD1的加成机制中涉及苄基自由基中间体的生成(见图4),因此自由基的稳定性可能对化合物活性具有一定影响。2015 年Pieroni 等[35]以Vianello等[13]设计合成的环丙胺α位取代的TCP化合物为骨架,在其α和β位分别引入影响苄基自由基稳定性的吸、供电子基,以考察对活性的影响。构效关系研究显示:在α位引入乙基取代所得的化合物21的活性明显提高,且同时在β位苯环间位上引入弱吸电子基的取代也有利于化合物活性增强,如在化合物21基础上引入β-苯环间位Br 取代,得到活性最好的化合物22(IC50=0.031μmol · L-1)。对接结果表明:化合物22与LSD1的2种结合模式(见图10)均异于骨架21(见图11),这种结合模式的变化可能更有利于化合物22与其周围残基的相互作用,从而导致活性增强。同时化合物HOMO-LUMO轨道能量差值△E计算结果表明化合物22的△E最低,可能更利于环丙烷的开环,因此活性最佳。
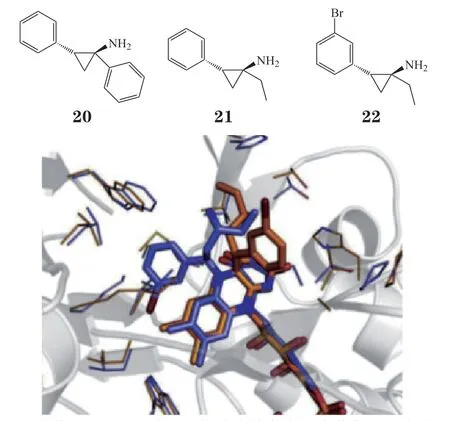
图10 化合物22-LSD1复合物的2种构象Figure 10 Two bindingcon formations of compound 22-LSD1
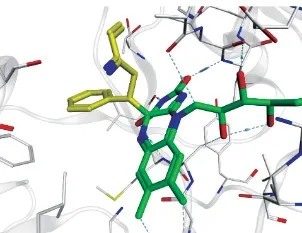
图11 化合物21-LSD1复合物构象Figure 1 1Complex conformation of compound21-LSD1
3 结语
LSD1作为一种新型抗肿瘤靶点,其抑制剂的研究开发成为热点,主要涉及三大类:1)基于底物的可逆性抑制剂;2)基于FAD的可逆性抑制剂;3)不可逆抑制剂。目前为止,TCP类不可逆性抑制剂是研究最深入、最全面的一类,以TCP为母体的结构改造得到了广泛研究。虽然TCP类抑制剂在多种癌症模型中表现良好,但是其缺乏LSD1特异性,会引发化合物脱靶效应,降低LSD1抑制效果的同时影响体内其他同源酶,引发副作用,因此有很多课题组致力于研发基于TCP的高选择性抑制剂。
本文针对已发表的复合物晶体结构、抑制机制以及TCP类抑制剂的研究加以归纳总结。笔者发现TCP 类抑制剂的苯环、氨基以及环丙基等基团均可进行结构修饰,结构改造的多样化也说明了TCP类抑制剂具有较大的改造空间。目前在对TCP的结构改造上,针对苯环进行改造的研究较多,通过改造苯环引入疏水基团、与周围疏水残基相互作用来提高抑制剂活性,或者引入大基团增加选择性,可为后续进一步改造LSD1抑制剂提供指导。另外,基于氨基和环丙基基团进行结构修饰的报道相对较少,但已报道的结构也都表现出较好的抑制效果,且目前进入临床研究的2个TCP类抑制剂均为氨基被修饰的化合物,进一步证实了氨基修饰的可行性,但这2处结构修饰可能存在一定合成难度、稳定性较差以及收率较低等问题,这些问题也是TCP类抑制剂改造的难点所在。目前关于TCP类抑制剂的改造大多局限于某一基团,后续的结构设计可以考虑将3处结构修饰相结合,最终获得的化合物对LSD1的抑制效果和生物活性可能表现更佳。
以上这些研究可以为此类抑制剂的进一步设计、优化提供重要指导作用,相信随着对LSD1研究的不断深入,将会有更多安全、有效的LSD1抑制剂得到开发。目前有2个TCP类不可逆抑制剂GSK2879552和ORY-1001已处于临床研究阶段,并有数个化合物正进行临床前开发研究,显示了LSD1是一个有潜力的靶标,也表明了TCP是优势结构,具有理想的开发前景,其多样化的结构改造对LSD1靶点的认知以及抑制剂用于药物研发具有重要意义[36-37]。
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
中学化学(2022年5期)2022-06-17 16:51:48
原子与分子物理学报(2021年2期)2021-03-29 07:30:46
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
高中数理化(2020年1期)2020-02-29 02:21:18
中成药(2018年7期)2018-08-04 06:04:18
中成药(2018年3期)2018-05-07 13:34:18
四川师范大学学报(自然科学版)(2018年2期)2018-04-28 02:21:08
池州学院学报(2015年3期)2016-01-05 01:13:04
天津科技大学学报(2015年2期)2015-08-09 01:40:42
