
一个B1型短指/趾畸形家系的临床特征及基因突变分析
2020-05-21 06:41王青青卞莎莎王圣然娄桂予秦利涛郝冰涛廖世秀
郑州大学学报(医学版) 2020年3期
王青青,卞莎莎,王圣然,娄桂予,王 鑫,秦利涛,郝冰涛,廖世秀
1)河南省人民医院(郑州大学人民医院)医学遗传研究所;河南省遗传性疾病功能基因重点实验室 郑州 450003 2)郑州大学医学科学院 郑州 450052
先天性短指/趾(brachydactyly,BD)畸形是指(趾)骨和(或)掌(跖)骨发育不良导致指(趾)短小、缺失或融合,是一种家族遗传性疾病。短指(趾)畸形也可伴有其他手/足部畸形,如并指(趾)畸形、多指(趾)畸形、缩小畸形[1]。与所有先天性畸形一样,短指畸形可以单独出现或作为复杂畸形综合征的一部分出现[2]。短指畸形可分为5种类型(A~E型),相同类型的短指表型差异可能很大[3]。B型短指畸形 (brachydactyly type B,BDB)是短指畸形中最严重的一种,可分为B1(MIM 113000)和B2(MIM 611377)2个亚型。B1型短指畸形(brachydactyly type B1,BDB1)主要临床特征包括食指到小指末梢发育不全,伴有指甲部分或完全缺失,拇指受累少,偶可见拇指/趾宽大畸形,脚也受到类似影响,但程度较轻,严重程度因人而异[4]。本研究报道了在河南地区发现的一个短指/趾畸形家系,先证者及其家系临床特征分析符合BDB1型。通过二代测序发现先证者受体酪氨酸激酶样孤儿受体2(receptor tyrosine kinase like orphan receptor 2,ROR2)基因存在一新发的移码突变:c.2290delG(p.A764fs*10)。现将结果报道如下。
1 对象与方法
1.1研究对象先证者,女性,31岁,汉族,以“短指/趾畸形”为主诉就诊于郑州大学人民医院遗传咨询门诊。该家系来自河南省,共24人,9人出现短指症状,符合常染色体显性遗传规律(图1)。签署知情同意书后,采集家系内5例患者和3例正常亲属外周血。3位患者行双手/足X线检查。本研究经郑州大学人民医院伦理委员会批准。
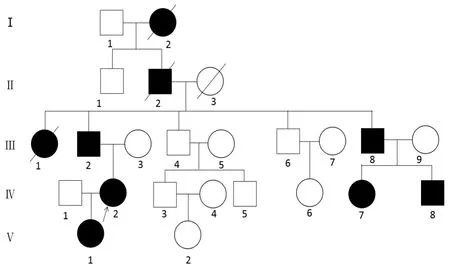
图1 短指/趾畸形家系图
1.2短指/趾畸形患者全基因组二代测序及基因突变分析使用EDTA抗凝管收集5例患者和3例正常亲属外周血3 mL,应用基因组提取试剂盒(QLAamp血液DNA纯化mini试剂盒)提取基因组DNA,使用Qubit dsDNA HS试剂盒对样品DNA进行定量及质量控制。按照Agilent公司SureSelectXT Target Enrichment System试验流程,采用捕获方法对2 742个基因(试剂编号:5190-7519)进行扩增,用Illumina Nextseq500二代测序仪对扩增产物进行测序分析。排除突变位点多态性后,发现先证者ROR2(NM_004560.3)基因上一个移码突变。针对该突变,使用Primer 5.0设计引物。ROR2上游引物序列5’-GCATTGGGATCTGCACCGGG-3’,下游引物序列5’-AAGGACCTGGCCACCCGCAA-3’;引物由上海生物工程技术服务有限公司合成。PCR扩增体系25 μL。PCR反应条件:95 ℃ 5 min变性;95 ℃ 30 s,64 ℃ 30 s,72 ℃ 1 min,共35 个循环;最后72 ℃延伸10 min。PCR扩增产物交由擎科生物有限公司进行Sanger双向测序验证。
2 结果
2.1该家系的临床特征该家系患者的手足外观检查见双手大拇指宽大扁平,第2~5指短小、末节指骨明显缩短或缺失,中、末节指骨关节处皮肤纹理消失、畸形指甲小而且发黑;所有患者足部均有受累,基本符合BDB1型表现(图2~4)。该家系患者的X线片显示双手大拇指末节指骨发育畸形,第2~5末端指骨缺失,部分中节指骨发育不良及中、末节指骨融合;双足X线片显示双足第2~5趾均有中节趾骨缺失,部分末节趾骨发育畸形(图2~4)。该家系患者的临床表现具有异质性,先证者Ⅳ2右手第3、5掌骨明显缩短,右足第4跖骨缩短,左足第3、5跖骨缩短(图2)。先证者父亲Ⅲ2双手外观检查见双手第2、3末节指骨缺失,第4、5中、末节指骨缺失。先证者堂弟、堂妹与先证者的父亲Ⅲ2表型相似(图3~4)。先证者叔叔Ⅲ8未参加本项研究。
2.2基因突变分析先证者Ⅳ2基因检测结果发现ROR2基因第9外显子上存在一个c.2290delG(p.A764fs*10)杂合突变(图5)。通过对先证者的父亲(Ⅲ2)、女儿(Ⅴ1)、堂弟(Ⅳ8)、堂妹(Ⅳ7)4位进行Sanger测序,发现均存在c.2290delG杂合突变。表型正常的Ⅲ3、Ⅲ4(图5)和Ⅳ5均无此突变。该致病突变在本家系中符合常染色体显性遗传规律(图1)。
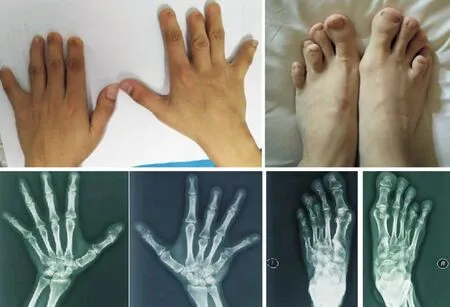
图2 先证者手足外观和X线片
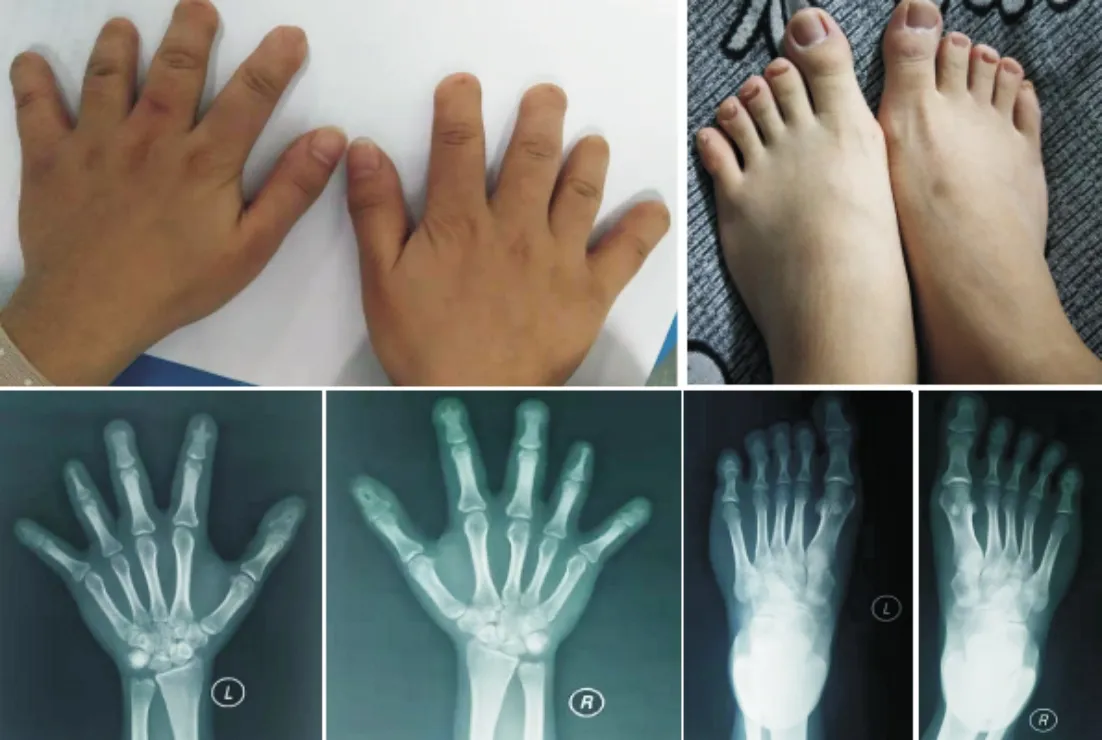
图3 先证者堂妹Ⅳ7手足照片及X线片

图4 先证者堂弟Ⅳ8及父亲Ⅲ2双手照片

图5 Ⅲ2、Ⅳ2、Ⅳ7、Ⅳ8、Ⅴ1患者以及Ⅲ4表型正常人的Sanger测序图
2.3基因突变功能预测该家系中ROR2基因c.2290delG杂合突变为移码突变,导致第764位密码子编码的氨基酸由丙氨酸转变为脯氨酸,且移码突变造成ROR2氨基酸序列提前终止,由正常编码的943个氨基酸变为773个氨基酸,致使该蛋白质部分功能区域缺失。
3 讨论
先天性手畸形是一种临床上高度异质性的疾病,临床表型差异非常大,包括并指、短指和缺指[5]。此外,一个特定的表型还包含几个亚型,如BDB1,这是一种常见的先天性手部畸形亚型,以指骨和指甲的远端发育不全为特征,部分患者伴有明显的面部体征,如人中短和鼻骨突出[6]。该家系所有患者以双手第2~5末端指骨短小或缺失,中、末节指骨融合,中节指骨发育不全为主要表现,基本符合BDB1的特点;同时该家系所有患者均有大拇指末节指骨畸形、中节趾骨缺失,部分患者存在掌骨(跖骨)缩短。BDB1也具有高度遗传异质性,但ROR2基因突变引起患者大拇指末节指骨畸形、中节趾骨缺失,部分患者存在掌骨(跖骨)缩短等表型少见报道。
ROR2基因定位于9q22,全长235 kb,共9个外显子,编码长度为943个氨基酸的ROR2蛋白[7]。ROR2基因编码的跨膜蛋白由胞外区、跨膜区、胞内区3个部分组成:与蛋白质-蛋白质相互作用相关的细胞外区域,包括免疫球蛋白样结构域(Ig)、卷曲的半胱氨酸富集区(CRD)和kringle结构域(Kr);胞内区域包括酪氨酸激酶域(TK)、2个丝/苏氨酸富集区(ST)和脯氨酸富集区(PRD)[8]。目前发现的BDB1致病基因多位于ROR2的8、9号外显子上,至今在中国人群中发现至少6种突变,多为无义突变或移码突变,导致ROR2蛋白胞内区TK附近的结构域缺失[9]。ROR2蛋白是Wnt5a的受体,其细胞外卷曲的CRD与Wnt5a结合,胞质区的PRD和S/TRD2与细丝蛋白A相结合,激活JNK而影响软骨细胞的增殖与极性迁移,该蛋白可能参与软骨细胞的早期形成,是软骨和生长板发育所必需的[9-10]。针对该家系的突变研究发现该突变产生一个只有774个氨基酸的截短ROR2蛋白,该截短蛋白缺少了ROR2蛋白的ST和PRD,可能导致Wnt5a/ROR2信号通路传导异常而引起BDB1。
本研究中,作者对一个符合常染色体显性遗传模式的BDB1型短指畸形家系进行了突变基因检测,发现该家系成员患病的遗传学基础是ROR2基因c.2290delG(p.A764fs*10)杂合突变。该突变与家系疾病表型共分离,查询dbSNP、人类基因突变数据库(HGMD)、ExAC外显子组整合数据库,均未见该变异报道,预测为新发现的致病突变,但需要进一步做功能验证。该突变丰富了ROR2突变谱,为进一步理解该疾病发病原因提供了遗传学基础,同时为该家系成员的遗传学咨询及产前诊断提供了分子依据。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
实用手外科杂志(2022年2期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
临床小儿外科杂志(2022年1期)2022-02-17
诊断学(理论与实践)(2020年1期)2020-04-28
郑州大学学报(医学版)(2019年3期)2019-06-03
中南林业科技大学学报(2019年4期)2019-04-08
森林工程(2018年1期)2018-05-14
中国医药科学(2016年2期)2016-10-09
