
UPLC-MS/MS法测定复方甘草片中甘草酸含量不确定度评定
2020-05-12 09:24:48陈高健周日秀汤丽昌通讯作者北海市食品药品检验所
食品安全导刊 2020年13期
□ 陈高健 周日秀 汤丽昌(通讯作者) 北海市食品药品检验所
复方甘草片为常用复方制剂,其成分包括甘草浸膏粉、阿片粉、樟脑、八角茴香油、苯甲酸钠等,属于镇咳祛痰类非处方药品[1]。UPLC-MS/MS具有较好的选择性和灵敏度,特别适用于复杂样品的分析。事实上,其测得结果只是被测量的估计值,而完整的测量结果还应包括被测量的估计值及不确定度[7,8]。因此,本文在建立UPLC-MS/MS法[2]测定复方甘草片中甘草酸含量的基础上,根据相关的规范和指南[3,4]并参考相关文献[5,6]对实验结果进行不确定度评定,以期为UPLC-MS/MS法测定复方甘草片中甘草酸的含量提供科学依据。
1 材料与方法
1.1 仪器与试剂
1290高效液相色谱仪,美国安捷伦公司;G6460三重串联四级杆质谱仪,美国安捷伦公司;S1-T256涡旋混合器;CPX5800H-C超声波清洗机,美国必能信上海科导仪器有限公司;XA205DU万分之一电子天平,上海梅特勒-托利多仪器有限公司。
甘草酸对照品(批号:G-004-170414)供实验室药品检验用,成都瑞芬思生物科技有限公司;复方甘草片(批号:190202)为市售品;色谱甲醇、色谱甲酸,Thermo;超纯水为Milipore 制备。
1.2 供试品溶液的制备
取复方甘草片20片,精密称定,研细后称取约1片量,置于100mL量瓶中,然后加入适量甲醇-水(1∶1),超声提取处理(300W,250KHZ)30min,取出后放冷;用甲醇-水(1∶1)稀释至刻度,摇匀,滤过,精密量取滤液1mL置于100mL容量瓶中;用甲醇-水(1∶1)稀释至刻度,摇匀后用0.22μm微孔滤膜滤过,取滤液即得标准供试品溶液,备用。
1.3 标准溶液的配制
精密称取甘草酸对照品15.67mg于50mL容量瓶中,加甲醇并摇匀,作为对照品储备溶液;精密吸取10mL储备液,置于100mL容量瓶中,加甲醇并摇匀,作为对照品标准溶液。精密吸取对照品标准溶液1.00、2.00、5.00、10.00、20.00mL,分别置于100mL容量瓶中,然后加甲醇并稀释至刻度,经0.22μm微孔滤膜过滤,供UPLC-MS/MS测试。
1.4 分析条件
Agilent EclipsePlus-C18色谱柱(2.1×50mm,1.8μm);流动相:0.1甲酸乙腈-0.1%甲酸水(70∶30,V/V);流速:0.2mL/min;柱温:30℃;进样量:5μL。采用电喷雾负电离源模式(Electronic spray ion,ESI-),扫描方式为多反应监测(Multiple reaction monitoring,MRM);雾化气压力:30psi;干燥气温度:300℃;干燥气流速:8L/min;毛细管电压:4000V;碎裂电压:165V;定量离子碰撞能:45eV,定性离子碰撞能:50eV。
1.5 测量模型
式中,X为样品中甘草酸的含量(mg),C为供试品溶液中甘草酸质量浓度(mg/mL),V为样品溶液的定容体积(mL),M为样品取样量(g)。
2 结果与分析
2.1 测量不确定度的来源
由测量模型可知,检测过程中产生的不确定度主要包括样品称量μrel(M)、样液定容μrel(V)、标准溶液配制μrel(C)、标准工作曲线拟合μrel(X)、样品重复性测试μrel(rep),以及加样回收率引入的标准不确定度μrel(R)。
2.2 各标准不确定度分量的评定
2.2.1 天平称取试样质量M引入的相对标准不确定度μrel(M)的评定
称取的试样质量为0.1500g。天平检定证书上的最大允许误差为±0.0001g,故由两次测量组成(称量空盘1次、盘加试样1次),按均匀分布,取包含因子 k=。
天平称取试样质量的标准不确定度为:
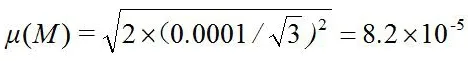
相对标准不确定度为:
μrel(M)=8.2×10-5/0.1500=5.5×10-4。
2.2.2 试样溶液制备的相对标准不确定度μrel(V)的评定
试样溶液定容由100mL容量和1mL单标吸量管产生,使用的两种容量器具引入的相对标准不确定度评定[7,8]结果见表1。
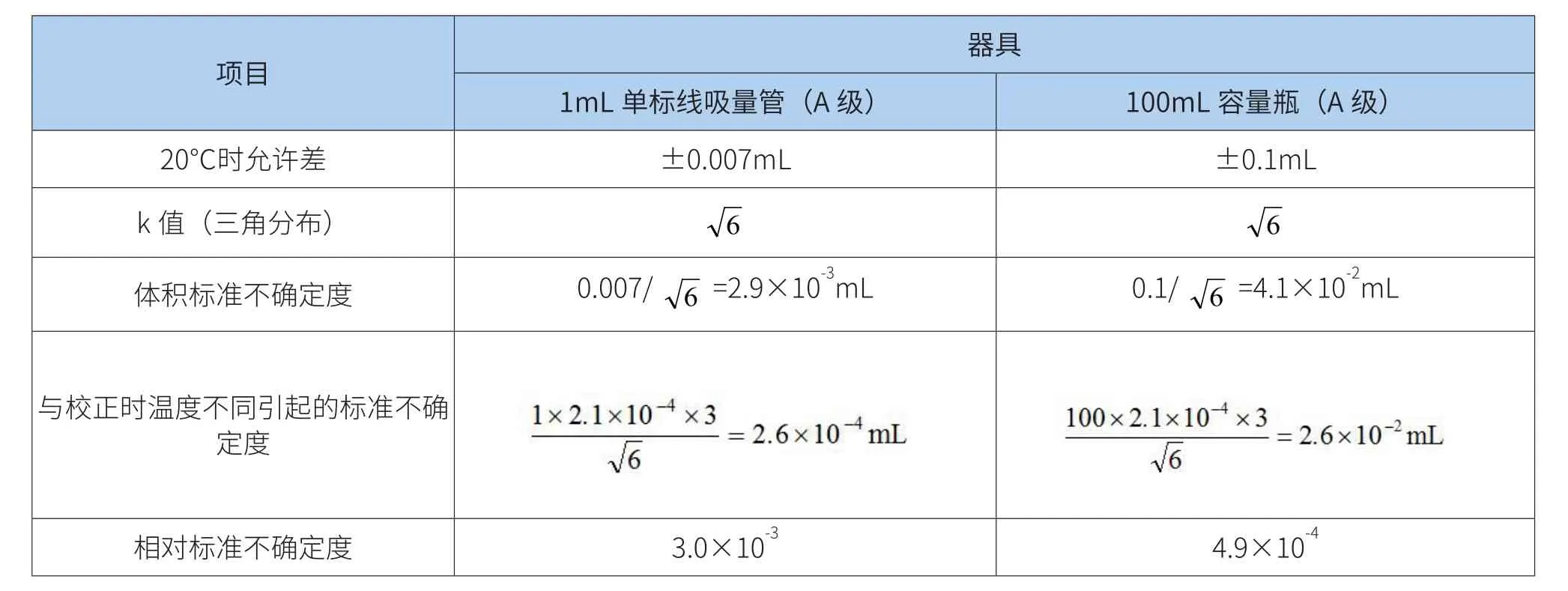
表1 试样溶液稀释引入的不确定度
注:水的膨胀系数为2.1×10-4℃-1。
样品稀释过程中引入的相对标准不确定度为:
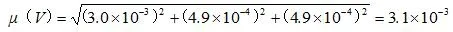
2.2.3 标准工作溶液配制过程中引入的相对标准不确定度μ1,rel(C)
2.2.3.1 标准物质引入的不确定度μ1,rel(C1)
测量所用标准物质甘草酸的纯度为98.0%,由标准物质证书得知相对标准不确定度为±2.0%,k=2,则μ1,rel(C1)=μ(Cs)/Cs=2%/2=1.0×10-2。
2.2.3.2 标准工作液稀释过程中引入的相对标准不确定度μrel(C2)
标准工作溶液不确定度由50、100mL容量和10mL单标吸量管产生,对所使用容量器具引入的相对标准不确定度评定[7,8]结果见表 2。
注:水的膨胀系数为2.1×10-4℃-1。
标准工作液稀释过程中引入的相对标准不确定度为:
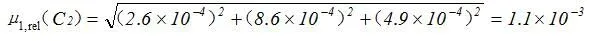
2.2.3.3 标准系列溶液配制过程中引入的相对标准不确定度 μ1,rel(C3)
吸取对照品溶液1.00、2.00、5.00、10.00、20.00mL,置于100mL容量瓶中,对所使用容量器具引入的标准不确定度评定[7,8]结果见表3。
注:水的膨胀系数为2.1×10-4℃-1。
标准系列溶液配制过程中引入的相对标准不确定度为:
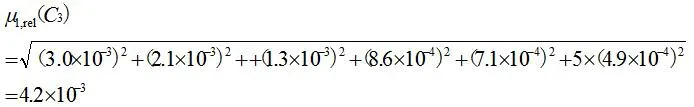
标准工作溶液配制过程中引入的相对标准不确定度为:

2.2.4 标准工作曲线拟合时引入的相对标准不确定度μrel(X)
根据工作站及采集数据对标准工作溶液进行测量,以响应值(A-)与浓度(C)作标准曲线得到拟合的线性回归方程:A=a+b×C=323.4×C+73.33,其中截距a=73.33,斜率b=323.4,相关系数r=0.9999。由工作曲线拟合时引入的不确定度源于其残差的标准差,残差为测定值(A)与标注曲线拟合值()之差的绝对值(|A-|),残差的计算见表4。


表2 标准品工作液稀释引入的不确定度
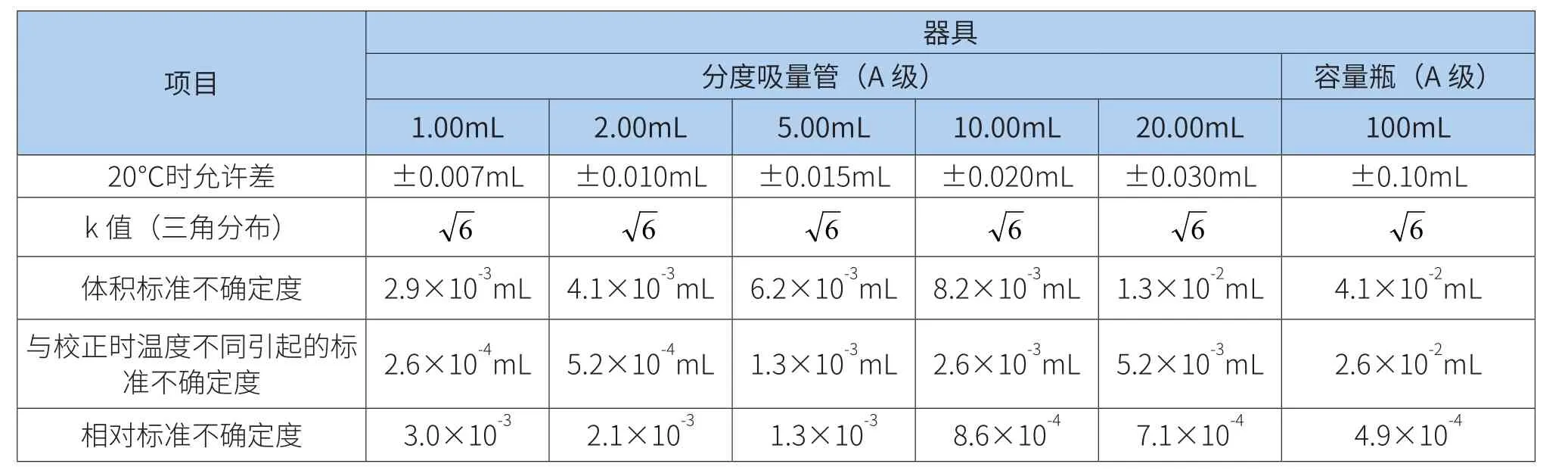
表3 标准系列溶液配制步骤引入的不确定度
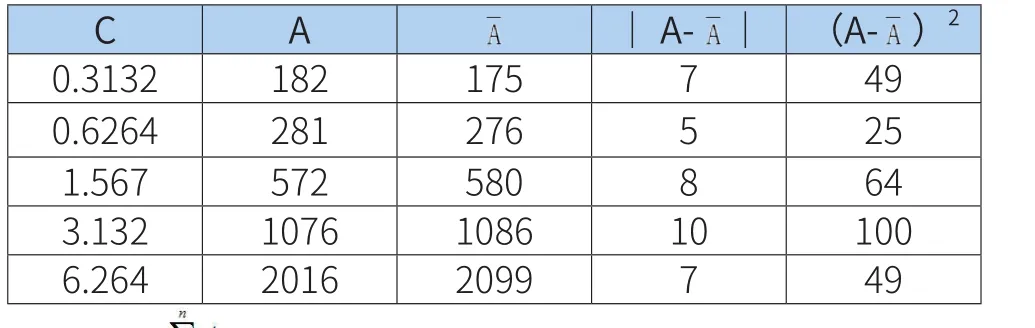
表4 标准工作曲线的残差计算结果
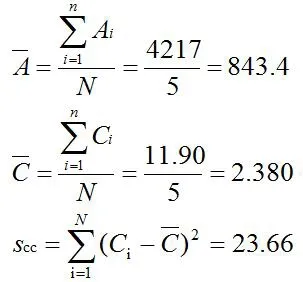
标准曲线残差标准差为:标准工作曲线拟合时引入的相对标准不确定度为:则甘草酸的扩展不确定度为Urel=2×3.9=7.8mg/g。因此,其真实结果可表示为(49.90±7.8)mg/g,k=2。

表5 各相对不确定度分量汇总表
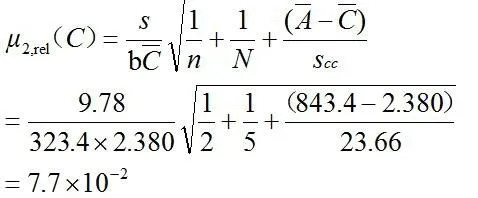
3 结论
由计算结果可知,本试验测量不确定度的主要来源是甘草酸标准溶液配制与标准曲线拟合。因此,需要重点考虑标准溶液的配制及标准系列的稀释过程中引入的不确定度分量。试验应严格按照操作规程,绘制好标准曲线,选择合理的线性范围,确保测量结果准确、可靠。经研究,本试验建立的不确定度评定方法可为UPLC-MS/MS测定复方甘草片中甘草酸含量提供一定的科学依据。
2.2.5 重复性实验引入的相对标准不确定度μrel(rep)的评定
取待测样品溶液,按试样方法分别进行6次测定,测定结果分别为49.87、50.23、48.98、50.39、51.11、48.79mg/g,平均值为49.90mg/g,标准偏差Srep=0.8823,重复性实验相对标准不确定度为
2.2.6 添加回收率引入的不确定度μrel(R)的评定
对样品进行6次加标回收试验,添加水平为0.1567μg/mL,回收率分别为100.2%、99.5%、98.1%、98.2%、97.3%和98.4%,平均回收率为98.6%,标准偏差为1.050,由回收率引入的相对不确定度为
2.3 甘草酸含量的合成标准不确定度和扩展不确定度
将各项相对标准不确定度分别列于表5,计算合成标准不确定度为:
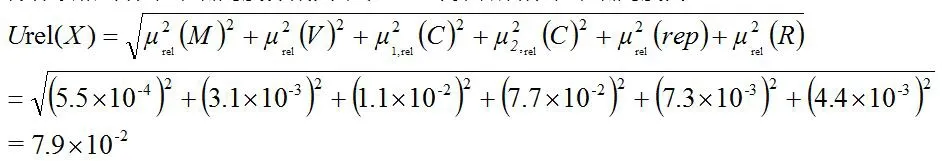
根据GB/T 27418-2017测量不确定度评定和表示法[3],采用UPLC-MS/MS测定甘草酸含量结果的合成不确定度为U(X)=49.90×7.9×10-2=3.9mg/g,取包含因子k(95)=2,
猜你喜欢
家庭医学(下半月)(2019年1期)2019-05-23 08:55:13
华声文萃(2018年11期)2018-07-13 19:25:21
益寿宝典(2018年14期)2018-01-27 18:53:04
价值工程(2017年31期)2018-01-17 00:34:27
电子测试(2017年12期)2017-12-18 06:35:46
水利科技与经济(2016年7期)2016-04-25 13:03:12
水利科技与经济(2016年8期)2016-04-22 03:41:22
中国卫生标准管理(2015年4期)2016-01-14 05:16:45
西南医科大学学报(2016年4期)2016-01-03 01:26:29
中国当代医药(2015年33期)2015-03-01 02:09:17
