
Neuroprotective mechanisms of ε-viniferin in a rotenone-induced cell model of Parkinson’s disease:significance of SIRT3-mediated FOXO3 deacetylation
2020-04-29 02:12:58ShuoZhangYanMaJuanFeng
中国神经再生研究(英文版) 2020年11期
Shuo Zhang, Yan Ma, Juan Feng,
1 Department of Neurology, Shengjing Hospital of China Medical University, Shenyang, Liaoning Province, China
2 Department of Ultrasound, Shengjing Hospital of China Medical University, Shenyang, Liaoning Province, China
Abstract Trans-(-)-ε-viniferin (ε-viniferin) has antioxidative and anti-inflammatory effects. It also has neuroprotective effects in Huntington’s disease by activating the SIRT3/LKB1/AMPK signaling pathway; however, it remains unknown whether ε-viniferin also has a neuroprotective role in Parkinson’s disease. A Parkinson’s disease cell model was induced by exposing SH-SY5Y cells to 3.0 μM rotenone for 24 hours, and cells were then treated with 1.0 μM ε-viniferin for 24 hours. Treatment with ε-viniferin upregulated SIRT3 expression, which promoted FOXO3 deacetylation and nuclear localization. ε-Viniferin also increased ATP production and decreased reactive oxygen species production.Furthermore, ε-viniferin treatment alleviated rotenone-induced mitochondrial depolarization and reduced cell apoptosis, and restored the expression of mitochondrial homeostasis-related proteins. However, when cells were transfected with SIRT3 or FOXO3 shRNA prior to rotenone and ε-viniferin treatment, these changes were reversed. The results from the present study indicate that ε-viniferin enhances SIRT3-mediated FOXO3 deacetylation, reduces oxidative stress, and maintains mitochondrial homeostasis, thus inhibiting rotenone-induced cell apoptosis. ε-Viniferin may therefore be a promising treatment strategy for Parkinson’s disease.
Key Words: deacetylation; FOXO3; mitochondrial homeostasis; mitophagy; oxidative stress; Parkinson’s disease; SIRT3; ε-viniferin
Introduction
Parkinson’s disease (PD) is one of the most common neurodegenerative diseases. Its etiology is not well understood, but is likely to involve both genetic and environmental insults,which may include exposure to specific pesticides (Freire and Koifman, 2012). With the development of global industrialization, the incidence of PD is trending upward, and the role of pollution and environmental factors is gaining importance (Chin-Chan et al., 2015). Furthermore, the progressive pathological changes that occur in PD are known to be caused by interacting mechanisms, including impairment of mitochondrial function, oxidative stress, abnormal α-synuclein protein aggregation, mitophagy, and apoptosis (Shefa et al., 2019).
Resveratrol is a polyphenolic compound naturally derived from several plants including grapes and Japanese knotweed,and it exerts a wide range of beneficial health effects by activating specific sirtuin family proteins (Tang, 2017). Previous studies have demonstrated the neuroprotective effects of resveratrol against neurotoxicity in a number of different PD models, including rotenone-exposed neuronal cells (Peng et al., 2016), a 6-hydroxydopamine (6-OHDA)-induced PD rat model (Jin et al., 2008), and a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated PD mouse model (Guo et al., 2016).However, although resveratrol has shown promise in the treatment and prevention of PD in a range of models, no clinical trials have confirmed the benefits of resveratrol in patients with PD. Moreover, when high-dose resveratrol was used over a long time period (corresponding to its use in animal models), low bioavailability (Smoliga and Blanchard,2014) and multiple adverse effects were reported in human studies (Cottart et al., 2014), including some serious adverse events (Heebøll et al., 2016).
Viniferin is the general name for resveratrol oligomers.Trans-(-)-ε-viniferin (ε-viniferin) is a dehydrodimer with a five-membered oxygen heterocyclic ring. Despite the structural similarities between resveratrol and ε-viniferin, these compounds modulate some neurobiological effects in different ways (Groundwater et al., 2015). Furthermore, ε-viniferin has been reported to have more effective antioxidant and anti-inflammatory activities than resveratrol (Zghonda et al.,2012). Fu et al. (2012) also demonstrated that low-dose viniferin has a neuroprotective effect on Huntington’s diseasein vitrovia activation of the SIRT3/LKB1/AMPK pathway.
SIRT3, a mitochondrial sirtuin, is characterized by its profile of deacetylase activity, which regulates oxidative stress damage and restores mitochondrial function by regulating mitochondria-related signaling pathways. SIRT3 may be a promising target in the treatment of neurodegenerative disorders (Brown et al., 2013; Lombard and Zwaans, 2014).Furthermore, a previous study suggests that SIRT3 has a neuroprotective effect, attenuating nigrostriatal degeneration by strengthening the scavenging capacity for mitochondrial free radicals (Liu et al., 2015).
The SIRT3 substrate fork headbox O3 (FOXO3) is a forkhead transcription factor that mediates several signaling pathways by activating multiple genes involved in energy metabolism, oxidative stress, proteostasis, apoptosis, cellular development and differentiation, metabolic processes,autophagy, and longevity (Morris et al., 2015; Martins et al.,2016). FOXO3 also plays a vital role in neurodegenerative diseases and is involved in PD progression. Pino et al. (2014)demonstrated that FOXO3 is a crucial factor for the survival of dopaminergic neurons, and may mediate the elimination of abnormal α-synuclein aggregation. Meanwhile, Tseng et al.(2013) reported that SIRT3 induced FOXO3 deacetylation and restored mitochondrial function by resisting oxidative stress and preserving mitochondrial function, and may be a possible target for the treatment of neurodegenerative diseases.
However, our understanding of the pathophysiological mechanisms of PD that are affected by SIRT3-induced FOXO3 deacetylation remains limited; furthermore, the therapeutic role of ε-viniferin in PD has not yet been completely elucidated. The purpose of the present study was to indentify whether ε-viniferin exerts a neuroprotective effect in response to rotenone-induced neurotoxicity by promoting the SIRT3-mediated FOXO3 deacetylation pathway in a cellular model of PD. In addition, SIRT3-induced FOXO3 deacetylation was also investigated to explore its effect on the expression of multiple mitochondrial homeostasis-associated proteins.
Materials and Methods
Cell culture and treatment
Human neuroblastoma SH-SY5Y cells lines were obtained from the Shanghai Institute of Biochemistry and Cell Biology.Cells were cultured in Dulbecco’s modified Eagle’s medium(DMEM)/F-12 supplemented with 100 U/mL penicillin/streptomycin and 20% fetal bovine serum. The cells were conventionally cultured and were maintained at 37°C in a humidified atmosphere of 95% sterilized air and 5% carbon dioxide.
Rotenone (10 mg; Aladdin, Shanghai, China) was diluted in 2 mL of 20% dimethyl sulfoxide. In addition, a stock of 12.7 mM rotenone was prepared for further use. For the toxicity experiments in SH-SY5Y cells, rotenone stock solutions were dissolved in phosphate-buffered saline (PBS). SH-SY5Y cells were plated in 96-well plates at a density of 1 × 104and exposed to various concentrations (500 nM, 1 μM, and 10 μM) of rotenone for 24 hours to create anin vitromodel of PD (Xicoy et al., 2017). Cell mortality following exposure to different concentrations of rotenone was measured using a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. The optimum concentration of rotenone was then chosen for further use.
Next, ε-viniferin (Wako Pure Chemical Industries, Osaka,Japan) was used as a therapeutic agent, and its ability to attenuate rotenone-induced neurotoxicity in the PD SH-SY5Y cell model was investigated. The intervention concentrations were 0-1.0 μM, and the intervention time periods were 0-48 hours. The optimum concentration of ε-viniferin was then chosen for further use.
Transfection and cell group assignment
To determine whether the SIRT3-mediated FOXO3 deacetylation pathway is necessary for the neuroprotective effect of ε-viniferin, short hairpin RNA (shRNA) transfection ofSIRT3,FOXO3, and control shRNA (MISSION®shRNA Plasmid DNA, Sigma-Aldrich, St. Louis, MO, USA)was performed. Briefly, cells were placed in six-well plates at 85-95% confluence. For the control cells, 10 μL of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was diluted with 250 μL of Opti-MEM Medium (Invitrogen). For the shRNA transfections, 250 μL of this Opti-MEM Medium/Lipofectamine 2000 mixture was added to each tube with 4 μg of shRNA and incubated for 20 minutes at room temperature. This mixture was then added to each well containing cells and medium. After being mixed by gentle rocking, the cells in the six-well plates were incubated at 37°C in a CO2incubator and then assayed for the target gene. Compounds were removed after 4 hours and medium was replaced. Cells in six-well plates at a density of 1 × 106were used for the following experiments. Six different treatment groups were designed, and the optimum exposure time and concentrations of ε-viniferin and rotenone were determined using the MTT assay. In the model group, cells were exposed to 3.0 μM rotenone for 24 hours, washed, and treated with 0.05%dimethyl sulfoxide (rather than ε-viniferin) for 24 hours. In the ε-viniferin treatment group, cells were first exposed to rotenone for the indicated time, and then 2 mL of 3.0 μM ε-viniferin was added to rescue the cells for 24 hours. In the ε-viniferin +SIRT3shRNA group, cells were transfected with 4 μg ofSIRT3shRNA prior to 3.0 μM rotenone exposure.In the ε-viniferin + FOXO3 shRNA group, cells were transfected with 4 μg of FOXO3 shRNA prior to 3.0 μM rotenone exposure. In the ε-viniferin + control shRNA group, cells were transfected with 4 μg of scrambled shRNA prior to 3.0 μM rotenone exposure. The control group was administered 0.05% dimethyl sulfoxide for the indicated time (the same as the model group).
Western blot assays were performed to test the protein levels of SIRT3 and FOXO3. After screening for the most effectiveSIRT3orFOXO3shRNA, the selected shRNA was used for the subsequent experiments. The SIRT3 expression level when inhibited by the selected shRNA was as high as 76% compared with controls; likewise, the FOXO3 expression level when inhibited by selected the shRNA was as high as 70% compared with controls (Additional Figure 1).
Cell viability test for rotenone exposure
Cell viability after rotenone exposure was tested using the MTT assay. This test is based on the ability of viable cells to convert soluble MTT into the insoluble, dark blue formazan(Kumar et al., 2018). For this assay, MTT (Beyotime, Shanghai,China) was dissolved in PBS at a concentration of 5 mg/mL and sterilized by passage through a 0.22 mm filter. The MTT reagent (10 μL; Beyotime) was added to each well after treating with different conditions in a 96-well plate, and the cells were incubated for 4 hours. Next, 100 μL of detergent reagent was added to each well and mixed thoroughly to dissolve the dark blue crystals. The plates were left at 37°C in the dark for 4 hours, and the absorbance at 570 nm was measured using a microplate reader (Bio-Rad, Hercules, CA, USA). The ratio of the absorbances recorded from the model and control groups was used to express cell viability.
Intracellular reactive oxygen species
Intracellular reactive oxygen species (ROS) generation in cells was evaluated by flow cytometry using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). Rotenone-induced neurotoxicity involves oxidative stress; specifically, rotenone is a mitochondrial complex I chain inhibitor (Pal et al.,2014). Intracellular ROS was measured using a ROS Assay Kit (Beyotime). DCFH-DA is oxidized by ROS in healthy cells to 2′,7′-dichlorofluorescein, which is highly fluorescent at 530 nm (Gu et al., 2018). Procedures were performed according to the manufacturer’s instructions. Briefly, SHSY5Y cells in six-well plates were washed three times with PBS. DCFH-DA, diluted to a final concentration of 10 μM,was added and incubated in the dark for 30 minutes at 37°C.After three washes with PBS, the relative fluorescence levels were recorded using flow cytometry (BD Biosciences, Mountain View, CA, USA) at excitation/emission wavelengths of 488/525 nm. Cells in the control group, treated with ROS only, were used as negative controls.
Mitochondrial membrane potential
Mitochondrial membrane potential was measured to determine whether the mitochondrial apoptotic pathway is associated with the neuroprotective mechanisms of ε-viniferin against rotenone-induced neurotoxicity. When JC-1 aggregates are taken up by mitochondria, they fluoresce red;this fluorescence requires a mitochondrial membrane potential. Monomeric green JC-1 remains in the cytosol, and the red/green ratio of JC-1 is an indicator of mitochondrial membrane potential. In brief, red fluorescence is increased(with weak green fluorescence) as a result of elevated mitochondrial membrane potential. A JC-1 Kit (Beyotime) was used to quantify mitochondrial membrane potential (Δψm).Procedures were performed according to the manufacturer’s instructions. Briefly, cells were seeded into six-well plates.The cells were incubated with the fluorescent, lipophilic,cationic probe JC-1 (5 μg/mL) for 20 minutes at 37°C and then washed twice with JC-1 staining buffer. The ratio of red(JC-1 aggregates) to green (JC-1 monomers) fluorescence intensities detected by flow cytometry (BD Biosciences) was considered as Δψm (Gu et al., 2018).
Reduced rotenone-induced mitochondrial depolarization and the neuroprotective effect of ε-viniferin were determined by the ratio of red/green fluorescence. The Δψm for each treatment group was also observed as the relative ratio of dual emissions from mitochondrial JC-1 green fluorescence (monomers) and red fluorescence (aggregates) using a fluorescent microscope (Nikon, Tokyo, Japan). Cell apoptosis induced by mitochondrial depolarization was measured using flow cytometry.
Intracellular ATP measurement
Intracellular ATP was assessed using an ATP Bioluminescence Assay Kit (Beyotime). Procedures were performed according to the manufacturer’s instructions. Briefly, cells in six-well plates were lysed in 200 μL of lysis buffer solution,followed by repeated pipetting and centrifugation at 4°C and 12,000 ×gfor 10 minutes. The supernatants were used for ATP detection. Protein concentrations were determined by BCA assay. For this assay, 50 μL of supernatant was added to 100 μL of ATP detection solution and incubated at room temperature for 5 minutes, and then mixed immediately.Luminescence was measured using a multi-mode microplate reader (BMG, POLARstar Omega, Offenburg, Germany).The ATP concentrations (nmol/μg protein) were obtained from an ATP standard curve (Tseng et al., 2013).
Apoptosis detection
An Annexin V-FITC Apoptosis Detection Kit with PI (Beyotime) was used to detect apoptosis after rotenone exposure.Procedures were performed according to the manufacturer’s instructions (Gu et al., 2018). Briefly, the assay was performed by analyzing FITC-labeled binding and PI uptake using the Annexin V-FITC Apoptosis Detection Kit. After treatment, SH-SY5Y cells from the different groups were collected and washed with PBS. Supernatant was removed after centrifuging for 5 minutes, and 195 μL of binding buffer and 5 μL of Annexin V-FITC were added. The cells were incubated for 10 minutes in the dark at room temperature before being centrifuged. After removing the supernatant, 190 μL of binding buffer and 10 μL of PI were added to the cell pellet.The cells were then incubated for 5 minutes in the dark at room temperature. After positioning the quadrants on the Annexin V/PI dot plots, live cells (Annexin V-/PI-), early/primary apoptotic cells (Annexin V+/PI-), late/secondary apoptotic cells (Annexin V+/PI+), and necrotic cells (Annexin V-/PI+) were distinguished. When calculating the total percentage of cells with fluorescence, both Annexin V+/PI-and Annexin V+/PI+cells were included. The fluorescence per sample was analyzed using flow cytometry (BD Biosciences).
Western blot assay
Western blot assays were performed to identify whether ε-viniferin affects mitochondrial homeostasis by indirectly regulating protein expression through the SIRT3/FOXO3 pathway.Procedures were performed according to the manufacturer’s instructions. Cells were washed with PBS and lysed in RIPA buffer containing protease and phosphatase inhibitors. After sonication, the suspension was centrifuged at 10,000 ×gat 4°C for 15 minutes, and the resulting protein concentration of the supernatant was determined using a BCA Protein Assay Kit (Beyotime). Western blot assays were performed with equal amounts of proteins separated on 5-12% Bis-Tris gels,and transblotted onto polyvinylidene difluoride membranes(Millipore, Billerica, MA, USA). Membranes ware blocked with 5% milk or 5% bovine serum albumin in Tris-buffered saline with 0.1% Tween 20 for 2 hours, and then incubated overnight at 4°C with primary antibodies against SIRT3(1:1000, 2627; Cell Signaling Technology, Beverly, MA, USA),FOXO3 (1:1000, sc-48348; Santa Cruz Biotechnology [SCBT],Santa Cruz, CA, USA), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α; a mitochondrial biosynthesis-related protein; 1:1000, sc-13067; SCBT),mitochondrial transcription factor A (TFAM; a mitochondrial biosynthesis-related protein; 1:1000, sc-30963; SCBT),DRP1 (a mitochondrial fusion and fission protein; 1:1000,sc-271583; SCBT), FIS1 (a mitochondrial fusion and fission protein; 1:1000, sc-98900; SCBT), MFN2 (a mitochondrial fusion and fission protein; 1:1000, PB0264; Boster, Wuhan,China), Parkin (a mitophagy-related protein; 1:1000, PB0342;Boster), NIX (a mitophagy-related protein; 1:1000, 12396S;CST), BNIP3 (a mitophagy-related protein; 1:1000, BA1302;Boster), and P62 (1:1000, PB0458; Boster). After being incubated at 37°C for 1 hour with appropriate horseradish peroxidase-conjugated goat antibodies to mouse or rabbit IgG(1:5000; Beyotime), the blots were developed using an ECLPlus Detection Kit (Beyotime). Densitometric analyses of the western blots were recorded using a Gel-Pro Analyzer (Media Cybernetics, Silver Spring, MD, USA) after scanning.
Immunoprecipitation and immunoblotting
To determine whether ε-viniferin enhances FOXO3 deacetylation by SIRT3, immunoprecipitation and immunoblotting were performed according to previous protocols (Tseng et al.,2013). Total protein isolated from cells (2 μg/μL) was incubated with mouse anti-FOXO3 (1:200, sc-48348; SCBT) at 4°C for 4 hours. Next, 50 μL of precleared protein G-Sepharose(Thermo Scientific, Carlsbad, CA, USA) was added and incubated at 4°C overnight. The resulting immunoprecipitate was boiled with 35 μL of sodium dodecyl sulfate reducing sample buffer and used for immunoblotting with mouse monoclonal anti-FOXO3 (1:200, sc-48348; SCBT), and rabbit anti-acetylated lysine (Ack; 1:50, ICP0380; ImmuneChem, Burnaby,BC, Canada). After being incubated at 37°C for 1 hour with appropriate horseradish peroxidase-conjugated goat antibodies to mouse (for FOXO3) or rabbit (for Ack) IgG, immunoprecipitations were performed using a Pierce Direct IP Kit(Thermo Scientific) according to the manufacturer’s instructions and western blot assays were conducted according to previous method (the section of western blot assay).
Subcellular fractionation
The biological activity of FOXO3 occurs via its nuclear relocalization in response to deacetylation, and as a transcription factor. The biological function of FOXO3 is mainly modulated by subcellular relocalization in response to external signals (Calnan and Brunet, 2008; Tseng et al., 2013).Subcellular fractionation and western blot assays were used to detect FOXO3 content in the cytosolic, nuclear, and mitochondrial fractions according to a previous protocol (Tseng et al., 2013). A BCA Protein Assay Kit (Beyotime) was used to measure the protein concentrations of the three fractions.Equal amounts of mitochondrial protein, nuclear protein,and cytosolic protein were then prepared. The western blot assays were conducted as described in the western blot assay section of our methods. Primary antibodies included mouse anti-FOXO3 (1:1000, sc-48348; SCBT), rabbit anti-VDAC1(1:2000, ab154856; Abcam, Cambridge, UK) as an internal control of mitochondrial proteins, mouse anti-Lamin A(1:500, ab8980; Abcam) as an internal control of nuclear proteins, and rabbit anti-β-actin antibody (1:1000, Beyotime) as an internal control of cytosolic proteins. After being incubated with appropriate horseradish peroxidase-conjugated goat antibodies to mouse or rabbit IgG (1:5000, Beyotime),the blots were performed using the ECL-Plus Detection Kit(Beyotime).
Immunofluorescence
Immunofluorescence was used to detect the expression levels of SIRT3 in the ε-viniferin treatment, control, and model groups. Briefly, after fixation at room temperature for 15 minutes with 4% paraformaldehyde, SH-SY5Y cells in sixwell plates were permeabilized with 0.2% Triton X-100, and then incubated at 4°C overnight with rabbit primary antibodies against SIRT3 (1:200, Cell Signaling Technology).After washing three times with PBS, the cells were incubated for 2 hours at 37°C with Alexa Fluor 568-conjugated goat-anti-rabbit secondary antibody (1:500; Invitrogen). To visualize nuclei, 10 μg/mL DAPI solution (Beyotime) was used. Images were obtained using a fluorescent microscope(Nikon).
Transmission electron microscopy
Transmission electron microscopy was used to observe mitochondrial morphology in the ε-viniferin treatment,control, and model groups. The procedure was conducted in accordance with our previous study (Xu et al., 2016). Briefly,cells were washed twice. After fixing, dehydrating, and embedding the cells, sections were stained with uranyl acetate and lead citrate and observed using a transmission electron microscope (Hitachi, Tokyo, Japan).
Statistical analysi s
All measurement data are expressed as the mean ± SD. Med-Calc Statistical Software version 15.8 (MedCalc Software bvba, Ostend, Belgium) was used for statistical data analysis.The one-way analysis of variance with Bonferronipost hoctest was performed for multiple-group comparisons and two-group comparisons. The repeated-measures analysis of variance was performed to explore overall differences in cell viability between the different groups at different exposure times and ε-viniferin concentrations. A value ofP< 0.05 was considered statistically significant.
Results
ε-Viniferin protects cells against rotenone-induced neurotoxicity via the SIRT3/FOXO3 pathway
The neuroprotective effect of ε-viniferin on rotenone-induced neurotoxicity was assessed using the MTT assay,which also represents the mitochondrial activities of succinate dehydrogenase. With different concentrations of rotenone exposure (500 nM-10.0 μM), there were dose-dependent decreases in SH-SY5Y cell viability. Twenty-four hours after different doses of rotenone exposure, cell viability was 79.29% for 500 nM, 64.22% for 1.0 μM, and 31.41% for 10.0 μM, compared with the control group. The 50% concentration of inhibition (IC50) was 3.02 μM, which was consistent with a preliminary study reporting rotenone concentration(Peng et al., 2016). A rotenone concentration of 3.0 μM was therefore chosen for the subsequent experiments.
To identify the optimum conditions of ε-viniferin treatment, cells in the ε-viniferin treatment groups were incubated at different concentrations (50 nM-10 μM) of ε-viniferin for different times (0-48 hours) after 3.0 μM rotenone exposure. Six different concentrations of ε-viniferin and five different treatment times were screened for their effects on viability using the MTT assay. As shown in Additional Figure 2A, after 3.0 μM rotenone exposure, specific concentrations of ε-viniferin significantly ameliorated cell viability in a time- and concentration-dependent manner. A peak at which the measured cell viability did not increase sharply occurred at a specific time (24-48 hours) and concentration(1-10 μM) (Additional Figure 2A and B). Thus, an ε-viniferin concentration of 1.0 μM and a treatment time of 24 hours were chosen for the subsequent experiments.
Treatment with ε-viniferin for 24 hours significantly improved cell viability after exposure to 3.0 μM rotenone, improving cell viability by 40.0 ± 1.8% compared with the model group (P< 0.05). The neuroprotective effect of ε-viniferin was significantly attenuated when cells were pre-transfected with eitherSIRT3orFOXO3shRNA. Compared with the ε-viniferin treatment group, the cell viabilities of the ε-viniferin +SIRT3shRNA group and the ε-viniferin +FOXO3shRNA group were significantly reduced, to 28.2 ± 2.0% and 20.3 ± 2.1%, respectively (P< 0.01). Furthermore, cell viability was not significantly different between the ε-viniferin treatment group and the ε-viniferin + control shRNA group(P= 0.43; Additional Figure 2C).
ε-Viniferin alleviates rotenone-induced mitochondrial depolarization
Compared with the model group, the mitochondrial membrane potential was markedly higher in the ε-viniferin treatment group (P< 0.05). To determine whether the SIRT3/FOXO3 pathway was involved in the reduction of mitochondrial depolarization by ε-viniferin, the effect ofSIRT3orFOXO3shRNA on ε-viniferin protection was tested. The mitochondrial membrane potentials of the ε-viniferin +SIRT3shRNA and ε-viniferin +FOXO3shRNA groups were significantly lower than that of the ε-viniferin treatment group(P< 0.05). Pre-transfection withSIRT3orFOXO3shRNA therefore abated the decrease in mitochondrial depolarization that was caused by ε-viniferin treatment. However, there was no significant difference in mitochondrial membrane potential between the ε-viniferin +SIRT3shRNA group and the ε-viniferin +FOXO3shRNA group (Figure 1A and B).Similarly, using flow cytometry, the apoptotic rate induced by mitochondrial depolarization was much higher in the model group than in the ε-viniferin treatment group. However, when SIRT3 or FOXO3 expression was suppressed, the protective effect of ε-viniferin was weakened (Figure 1C).
ε-Viniferin decreases rotenone-induced oxidative stress
After rotenone exposure, the production of intracellular ROS was higher in the model group than in the control group.When cells were treated with ε-viniferin for 24 hours, ROS levels were significantly lower compared with the model group (P< 0.05). In addition, the ROS levels as determined by fluorescence were higher in the ε-viniferin +SIRT3shRNA or ε-viniferin +FOXO3shRNA groups compared with the ε-viniferin treatment group (P< 0.05). These results suggest that ε-viniferin may reduce oxidative stress via the SIRT3/FOXO3 pathway (Figure 2A and B).
ε-Viniferin increases ATP production in rotenone-treated SH-SY5Y cells
The effect of ε-viniferin on cellular ATP production was evaluated in SH-SY5Y cells. Compared with the control group, ATP production was markedly lower in SH-SY5Y cells that were exposed to rotenone (Figure 2C). Compared with the model group, cellular ATP production was restored when cells were treated with ε-viniferin (P< 0.05). However,in cells that were transfected withSIRT3orFOXO3shRNA prior to ε-viniferin treatment, this increase in ATP production was reversed (P< 0.05). Therefore, ε-viniferin may promote ATP production in cells exposed to rotenone via the SIRT3/FOXO3 pathway.
ε-Viniferin mitigates rotenone-induced apoptosis
Figure 3 shows a dot-plot display yield by Annexin V-FITC/PI using flow cytometry. After 24 hours of rotenone exposure, 53.7% of SH-SY5Y cells were apoptotic. However, incubation for 24 hours with ε-viniferin reduced the percentage of apoptotic cells to 24.2% compared with the model group(P< 0.05). When cells were pre-transfected withSIRT3orFOXO3shRNA, the protective effect of ε-viniferin was abated. Moreover, there was no significant difference in apoptotic rates between the ε-viniferin +SIRT3shRNA group and the ε-viniferin +FOXO3shRNA group (P> 0.05). Therefore,ε-viniferin may suppress rotenone-induced apoptosis via the SIRT3/FOXO3 pathway.
ε-Viniferin promotes SIRT3-mediated FOXO3 deacetylation and nuclear localization
Next, we examined whether ε-viniferin also altered SIRT3 deacetylase activity by measuring acetylated FOXO3. Our findings indicated that ε-viniferin reduced acetylated FOXO3 levels, implying that ε-viniferin may ameliorate rotenone-induced SIRT3 decline and increase SIRT3-mediated FOXO3 deacetylation.
The FOXO3 expression in the different groups was separated into mitochondrial, nuclear, and cytosolic fractions.FOXO3 expression in each of the different groups was varied in all three of the subcellular compartments, and the ε-viniferin treatment group had distinct distributions. There was no contamination of cellular fractionation with any other markers. ε-Viniferin treatment significantly increased the nuclear localization of FOXO3 compared with the model group (53.6%vs. 10.50% in the nuclear fraction) (P< 0.05).These results indicate that ε-viniferin promotes SIRT3-mediated FOXO3 deacetylation and its nuclear relocalization(Figure 4).
ε-Viniferin drives the expression of mitochondrial homeostasis-related proteins by strengthening SIRT3 to mediate FOXO3 deacetylation
To determine whether PGC-1α is modulated by ε-viniferin via SIRT3-mediated FOXO3 deacetylation, cells were transfected with eitherSIRT3orFOXO3shRNA before ε-viniferin treatment. Rotenone-induced inhibition of PGC-1α was reversed after treatment with ε-viniferin; that is, PGC-1α levels were enhanced following ε-viniferin treatment. However, PGC-1α levels were sharply reduced when cells were pre-treated withSIRT3orFOXO3shRNA. ε-Viniferin also enhanced TFAM expression levels. These findings indicate that ε-viniferin promotes FOXO3 deacetylation by SIRT3,which then increases PGC-1α and TFAM expression during rotenone exposure. Our results also suggest that rotenone exposure induces an inhibition of DRP1, FIS1, and MFN2 expression, and that treatment with ε-viniferin can reinforce the biological activity of mitochondrial fission and fusion by promoting DRP1, FIS1, and MFN2 expression.The increased expression of these proteins was reversed by inhibiting SIRT3 or FOXO3 expression. Thus, our results suggest that ε-viniferin enhances mitochondrial fission and fusion by upregulating the SIRT3-mediated deacetylation of FOXO3. Our results also revealed that ε-viniferin treatment promotes the expression of NIX, BNIP3, and Parkin.When SIRT3 or FOXO3 activity was inhibited by shRNA,NIX, BNIP3, and Parkin expression levels were decreased compared with the ε-viniferin treatment group. ε-Viniferin pretreatment reversed the inhibition of SIRT3 and FOXO3 activation. Moreover, as an important marker for autophagy (Jiang and Mizushima, 2015), P62 is notably degraded during autophagy. ε-Viniferin treatment resulted in reduced P62 levels, while knockdown of SIRT3 and FOXO3 reversed the ε-viniferin-mediated suppression of P62 (Figure 5).
To determine the role of ε-viniferin in maintaining mitochondrial homeostasis, mitochondrial morphology was examined during ε-viniferin treatment using transmission electron microscopy. As shown in Figure 6, autophagosomes, autophagic compartments, and elongation of mitochondria were markedly increased in the ε-viniferin treatment group compared with the control group, or model group. Compared with the ε-viniferin treatment group, autophagosomes, autophagic compartments, and mitochondrial elongation were noticeably decreased in control group, and model group. Our results suggest that ε-viniferin promotes mitophagy flux by upregulating SIRT3-mediated deacetylation of FOXO3. These results suggest that ε-viniferin ameliorates rotenone-induced mitochondrial dysfunction.
Discussion
This study demonstrated that ε-viniferin is a novel stabilizer of mitochondrial function, and that it has a potential neuroprotective effect by promoting the SIRT3-mediated FOXO3 deacetylation pathway in response to rotenone-induced neurotoxicity in a cellular model of PD, thereby preserving mitochondrial transmembrane potential and ameliorating rotenone-induced oxidative stress injury. Moreover,SIRT3-mediated FOXO3 deacetylation enhances the expression of multiple mitochondrial homeostasis-related proteins,promoting TFAM and PGC-1α to coordinate mitochondrial biogenesis, activating MFN2, FIS1, and DRP1 to regain mitochondrial fission-fusion balance, and upregulating Parkin,BNIP3, and NIX to induce mitophagy in response to rotenone-induced neurotoxicity (Westermann, 2010; Archer,2013). The SIRT3-mediated FOXO3 deacetylation pathway is required for the neuroprotective mechanisms of ε-viniferin,because SIRT3 inhibition abolished these neuroprotective effects.
ε-Viniferin has been reported to have antioxidant and cytoprotective activities in Alzheimer’s disease (Vion et al.,2018; Caillaud et al., 2019). Early treatment with mitochondrial antioxidants may interfere with oxidative stress damage and rescue lysosomal dysfunction in PD, which highlights the importance of early therapeutic intervention in the pathogenic cascade (Burbulla et al., 2017). However, no previous studies have evaluated the therapeutic significance of ε-viniferin in PD or described the molecular mechanisms underlying its therapeutic effects. Our study indicates that ε-viniferin has a potent antioxidative effect and maintains mitochondrial homeostasis by activating SIRT3-mediated FOXO3 deacetylation; moreover, ε-viniferin also inhibits rotenone-induced apoptosis and downregulates ROS production in rotenone-treated SH-SY5Y cells. ε-Viniferin is thought to be a SIRT3 activator and may bind to SIRT3 protein. ε-Viniferin may thus be a therapeutic multi-target candidate with multiple pharmacological effects, rescuing both cytotoxic protein aggregation and oxidative stress damage in the treatment of PD. Our results also indicate that a detectable SIRT3 increase may be the consequence of mitochondrial depolarization after rotenone exposure, suggesting that SIRT3 may have a reflexive compensatory response against mitochondrial dysfunction.
Although it is well known that SIRT1 activation is the main mechanism underlying resveratrol-mediated anti-aging effects, it has been reported that resveratrol can also activate FOXO3 directly, without it being deacetylated by sirtuins, in pathological conditions (Franco et al., 2014;Kwon et al., 2017). Pre-treatment with a SIRT3 inhibitor interfered with the neuroprotective effects of ε-viniferin in a rotenone-induced cell model of PD. Our study did not identify that SIRT3 and FOXO3 are simultaneously activated by ε-viniferin; however, FOXO3 inhibition did not fully inhibit the protective effect of ε-viniferin, suggesting that ε-viniferin-induced SIRT3 activation might enhance other pathways for protecting mitochondrial function.
PD is characterized by multiple cellular and molecular damage mechanisms. Epidemiological studies have repeatedly identified that an increased risk of sporadic PD is associated with exposure to environmental contaminants such as pesticides, and many of these environmental toxins can be used to recapitulate PD pathology in animal and cell models(Goldman, 2014). In the present study, we used rotenone as a PD-causing neurotoxic agent. Rotenone works by interfering with the electron transport chain in mitochondria, resulting in the inhibition of the mitochondrial respiratory chain complex I. Cellular oxygen is then reduced to radicals, which can cause high oxidative stress, thereby leading to the selective degeneration of striatal and nigral dopaminergic neurons in mammals (Johnson and Bobrovskaya, 2015; Cao et al., 2019). A previous study summarized transcriptome data from SH-SY5Y cells after rotenone exposure, and demonstrated a pleiotropic reaction to rotenone exposure that causes a large range of molecular biological effects (Huo et al., 2012). In addition, a National Institutes of Health study reported a strong positive association between rotenone use and PD in farm workers (Tanner et al., 2011). Therefore, the significance of rotenone-induced neurotoxicity has provided new insights into the pathophysiological mechanisms of PD,and may lead to new therapeutic strategies.
An accumulation of defective mitochondria is mainly responsible for ROS production. Oxidative stress has been implicated in many disease processes that are mediated by mitochondria, especially in the pathogenesis of neurodegenerative diseases (Rimessi et al., 2016). Mitochondrial dysfunction is widely regarded to have a pivotal role in dopaminergic neuronal susceptibility, which is the characteristic feature of both the familial and sporadic forms of PD, as well as of neurotoxin-induced parkinsonism (Ryan et al., 2015). Targeting mitochondrial homeostasis might be a therapeutic strategy for PD (Shefa et al., 2019). Toxic insults may disrupt mitochondrial dynamics and result in the gradual degeneration and loss of dopaminergic neurons in the substantia nigra pars compacta, and the subsequent reduction of dopamine levels in the striatum. The maintenance of mitochondrial homeostasis relies on the dynamic balance between mitochondrial fusion and fission (biogenesis) as well as degradation (mitophagy), and other processes such as intracellular signaling pathways are also a part of this balance. Mitochondrial homeostasis is considered to be very important in PD pathogenesis. Likewise, the regulation of mitochondrial homeostasis is a promising target for developing new therapeutic strategies in PD. The PGC-1α, DRP1/FIS1/MFN2, and Parkin pathways coordinate a balance that regulates mitochondrial biogenesis, fission/fusion, and mitophagy (Peng et al., 2019). A defective balance in the molecular biological processes that induce mitochondrial biogenesis and selective mitophagy may cause potential disturbances in mitochondrial homeostatic dynamics (Freire and Koifman,2012). PGC-1α is known as a transcriptional coactivator and is vital for mitochondrial biogenesis (Kuo et al., 2012; Peng et al., 2016). TFAM is a key activator of mitochondrial transcription, as well as a downstream protein of PGC-1α.
Mitochondrial homeostasis is also determined by the dynamics of mitochondrial fission and fusion. The proteins regulating mitochondrial fission and fusion, DRP1 and FIS1,are indispensable components of a mitochondrial complex that promotes mitochondrial fission, while MFN2 is reported to be essential for mitochondrial fusion (Westermann,2010; Archer, 2013). Mitophagy, as a selective autophagic process, is responsible for the degradation of defective mitochondria following damage or stress in neurodegenerative diseases, and maintains mitochondrial function and integrity when cells are exposed to mitochondrial toxins (Vives-Bauza and Przedborski, 2011; Ashrafi and Schwarz, 2013). BNIP3,NIX, and Parkin are key regulators of mitophagy, and their expressions are positively regulated by FOXO3 during oxidative stress (Tseng et al., 2013; Li et al., 2015).
Mitochondria-related acetylation is also essential in the regulation of mitochondrial homeostasis. The SIRT3-mediated deacetylation of FOXO3 contributes to mitochondrial homeostasis in rotenone-induced neurotoxicity. To further confirm the molecular mechanisms underlying the neuroprotective effects of ε-viniferin, our study demonstrated that the activation of SIRT3-mediated FOXO3 deacetylation is induced by rotenone neurotoxicity and regulated by ε-viniferin neuroprotection. SIRT3 is a mitochondrial sirtuin that plays an important role in mitochondrial function. A previous study (Fu et al., 2012) confirmed that ε-viniferin significantly increased the levels of the 28-kDa active isoform in a Huntington’s disease cell model. In support of this previous study, we revealed that ε-viniferin-activated SIRT3 elevated the expression of mitochondrial homeostasis-related proteins. In the present study, activated SIRT3 maintained mitochondrial homeostasis against neurotoxin-induced oxidative stress by deacetylating FOXO3. In a previous study,the mitochondrial deacetylase SIRT3 was reported to initiate mitochondrial degradation by mitophagy in toxin-induced models of PD before the onset of PD-associated aggregate formation (Auburger et al., 2014). Our observations are in agreement with these earlier results, and demonstrate that SIRT3 levels increase markedly after rotenone exposure, as a negative feedback mechanism that has a protective effect.SIRT3/FOXO3 deacetylation is involved in mitochondrial biogenesis and is considered to be an important compensatory mechanism that allows adaptation to mitochondrial dysfunction caused by rotenone-induced neurotoxicity.However, SIRT3 protein levels may decline in response to persistent mitochondrial dysfunction. In general, ε-viniferin enhances the maintenance of mitochondrial homeostasis and diminishes oxidative stress and neurodegeneration in rotenone-induced neurotoxicity.

Figure 1 ε-Viniferin alleviates rotenone-induced mitochondrial depolarization.
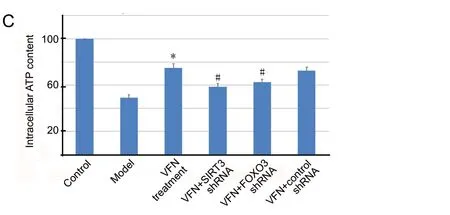
Figure 2 ε-Viniferin decreases rotenone-induced oxidative stress and increases ATP production.

Figure 3 ε-Viniferin mitigates rotenone-induced apoptosis.
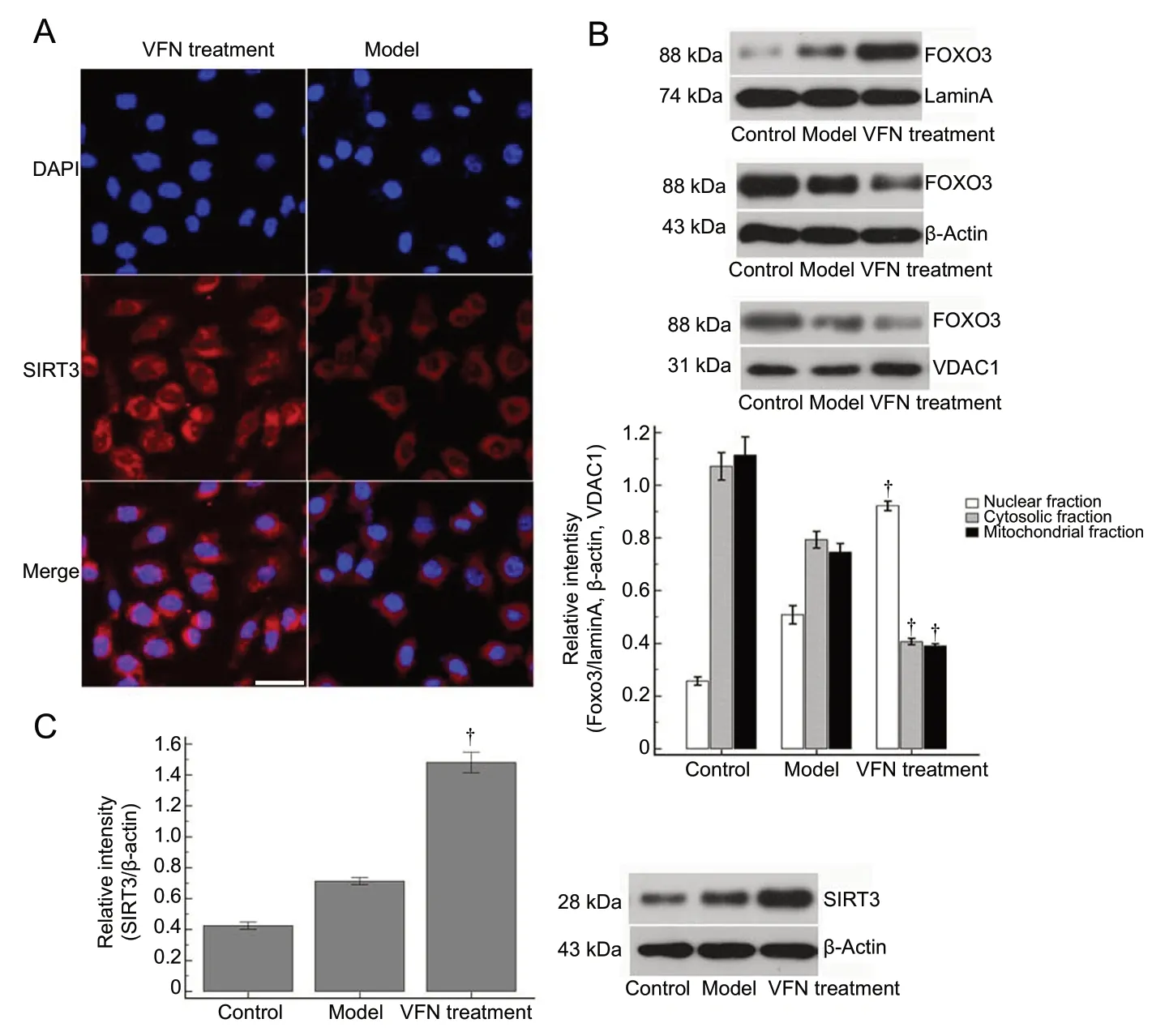
Figure 4 FOXO3 deacetylation induced by ε-viniferin promotes SIRT3 expression and FOXO3 nuclear localization.
SIRT3 deficiency is implicated in mitochondrial dysfunction. The Krebs cycle is a stage in the process of cellular respiration, and allows for the three major nutrients to be metabolized. Citrate synthase is a key mitochondrial enzyme in the Krebs cycle. Cui et al. (2017) suggested that high expression of SIRT3 can induce deacetylation of citrate synthase, activate citrate synthase, and increase ATP synthesis in an MPP-induced PD cell model. SIRT3 thus has a neuroprotective effect by deacetylating downstream targets and activating related mitochondrial enzymes. Moreover,Zhang et al. (2016) reported that SIRT3 knockdown in a rotenone-induced PD model significantly worsened the rotenone-induced decline in cell viability, enhanced cell apoptosis, exacerbated the decrease in antioxidant enzymes(superoxide dismutase and glutathione), reduced mitochondrial membrane potential, and rapidly increased α-synuclein accumulation. SIRT3 overexpression dramatically reversed these adverse effects. Meanwhile, SIRT3 has been demonstrated to mediate autophagic effects by strengthening LKB1 phosphorylation, leading to increased AMPK activation and decreased mTOR phosphorylation (Zhang et al., 2018), suggesting that SIRT3 regulates autophagy not only via FOXO3 acetylation, but also via other signaling pathways.
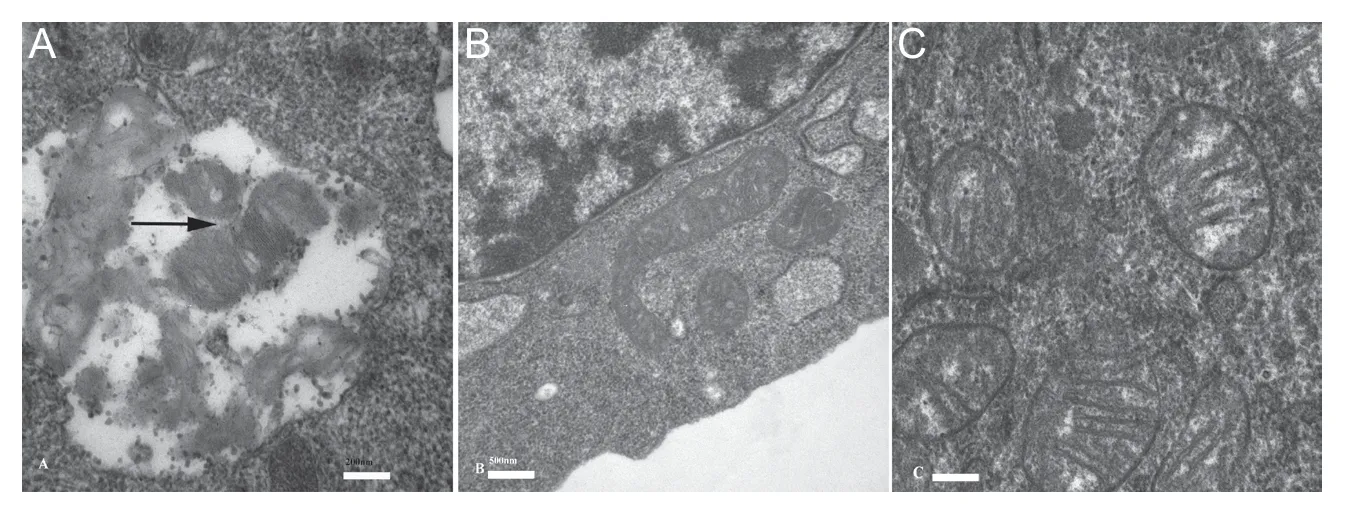
Figure 6 Mitophagy was observed by transmission electron microscopy.
Our study has some unavoidable limitations, which need to be addressed in future research. First, although there are limitations to experiments with neuroblastoma cell lines,the use of SH-SY5Y cells as a PD cell model provides a simple and convenient way to screen the antioxidant potentials of various natural compounds and pharmaceuticals. Our results should be verified in further studies using primary cultured animal dopaminergic neurons or human induced pluripotent stem cell-derived dopaminergic neurons, in the context of dopaminergic neurodegeneration. Second, the emerging role of autophagy in disease models using tumor cells is a double-edged sword. In the present study, ε-viniferin-induced autophagy enabled SH-SY5Y cells to tolerate oxidative stress from rotenone-induced cytotoxicity, at least to some extent. However, autophagy has been reported to play a vital role in damage mitigation that can limit tumorigenesis. In the present study, we did not investigate the effects of autophagy on the proliferation or invasion of SH-SY5Y cells without any treatment. Further studies are therefore needed to explore the effects of autophagy in the establishment of an SH-SY5Y PD cell model. Third, macroautophagic flux is another important neuroprotective mechanism against PD.Our results revealed an overexpression of mitophagy-related proteins with ε-viniferin treatment. Thus, our results indirectly suggest that mitophagy may be activated by ε-viniferin.However, further studies are needed using macroautophagic flux experiments with bafilomycin to determine whether ε-viniferin can promote macroautophagic flux. Finally, our PD cell model indicates that ε-viniferin may be clinically beneficial for treating PD. It is noteworthy that resveratrol treatment has no consistent clinical effect, even though resveratrol has been reported to have a slight ameliorating effect in somein vitroexperiments. Bioavailability may be an important factor limiting its clinical application. There has been no research into the bioavailability of ε-viniferin in animal models or in humans. In addition, adverse events with clinical ε-viniferin treatment should be paid attention.Further studies should explore the therapeutic effects of ε-viniferin on PDin vivo.
Conclusions
SIRT3/FOXO3 activation by ε-viniferin has neuroprotective effects in a rotenone-induced PD cell model, resulting from the increased expression of multiple proteins. The SIRT3-mediated FOXO3 deacetylation pathway is indispensable for mitochondrial homeostasis, including the promotion of mitochondrial biogenesis, the activation of mitochondrial fission/fusion, and the induction of mitophagy. Regulation of mitochondrial homeostasis might be a crucial therapeutic target for PD, and ε-viniferin may play a vital role in protecting mitochondria from neurotoxic injury via the SIRT3-mediated FOXO3 deacetylation pathway. Our study revealed that the activation of SIRT3 by ε-viniferin is a mechanism of ε-viniferin-induced mitochondrial homeostasis. The new insights from our study suggest novel mechanisms and therapeutic targets in PD.
Author contributions:Study concept and design and paper writing: SZ and JF; experimental implementation: SZ and YM. All authors reviewed and edited the paper, read and approved the final version of the paper.
Conflicts of interest:The authors declare that they have no competing interests.
Financial support:This work was supported by the National Natural Science Foundation of China, Nos. 81771271 (to JF), 81801710 (to YM);the Science and Technology Project Funds from Education Department of Liaoning Province of China, Nos. LK2016022 (to SZ), LK2016021 (to YM). The funding sources had no role in study conception and design,data analysis or interpretation, paper writing or deciding to submit thispaper for publication.
Institutional review board statement:SH-SY5Y cells are commercial products, and are not directly from humans or animals. So the study was not involved in ethical review.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:Additional Figure 1:The screening the most effective shRNA for inhibiting the protein expression of SIRT3 (A) and FOXO3 (B).Additional Figure 2:Neuroprotective effects of ε-viniferin on cell viability in a Parkinson’s disease model as detected by 3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide assay.
- 中国神经再生研究(英文版)的其它文章
- Muscovite nanoparticles mitigate neuropathic pain by modulating the inflammatory response and neuroglial activation in the spinal cord
- Knocking down TRPM2 expression reduces cell injury and NLRP3 inflammasome activation in PC12 cells subjected to oxygen-glucose deprivation
- Amyloid-beta peptide neurotoxicity in human neuronal cells is associated with modulation of insulin-like growth factor transport, lysosomal machinery and extracellular matrix receptor interactions
- MicroRNA regulatory pattern in spinal cord ischemiareperfusion injury
- Sequencing analysis of matrix metalloproteinase 7-induced genetic changes in Schwann cells
- Intraoperative single administration of neutrophil peptide 1 accelerates the early functional recovery of peripheral nerves after crush injury