
小立碗藓PpCASLUU3基因敲除载体的构建
2020-04-22 06:25:18闫慧清曹海鹏辛健康
西南农业学报 2020年1期
陈 波,闫慧清,李 莉,曹海鹏,辛健康,姜 山
(贵州师范大学 生命科学学院,贵州 贵阳 550001)
【研究意义】小立碗藓(Physcomitrellapatens)是葫芦藓目(Funariales)葫芦藓科(Funariaceae)小立碗藓属(Physcomitrium)的藓类。苔藓植物是非维管束植物,隶属于高等植物中较低等的类群,是最早登陆的陆生植物代表[1-3]。细胞凋亡(apoptosis)指为维持内环境稳定,由基因控制的细胞自主有序的死亡。涉及一系列基因的激活、表达以及调控等的作用,并不是病理条件下自体损伤的一种现象,而是为更好地适应生存环境而主动争取的一种死亡过程。半胱氨酸蛋白水解酶(cysteinyl aspartate specific proteinas, caspase)作为细胞凋亡过程的重要蛋白酶在细胞凋亡的最后实施过程中起决定性的作用,作为多种凋亡程序启动的执行者,有着非常重要的意义和作用[4]。其对底物的切割使得细胞呈现出凋亡的一系列形态和分子生物学特征:如形成凋亡小体、DNA Ladder、凋亡细胞皱缩等[5-7]。基因敲除技术是研究基因功能的基本手段,Caspase基因功能的研究拟采用基因敲除的技术,而基因敲除技术中载体的构建尤为重要。小立碗藓基因组已测序完成,其核基因组易于与有同源片段的外源DNA发生高频率的同源重组,从而使得精确的基因敲除成为可能,为基因功能的研究提供了良好的材料。目前已成为国内外植物分子生物学研究的模式生物[18]。【前人研究进展】虽然植物中没有Caspase的同源蛋白,但是植物的细胞程序性死亡(programmed cell death,PCD)过程有时能检测到Caspase的活性,这种酶活性被定义为类Caspase活性(caspase-like activity)[7],命名为类半胱氨酸蛋白酶(Metacaspase)[9],研究显示,有些植物能够产生类似Caspase活性的物质,并且这种酶活性能调控PCD过程[10]。在拟南芥(Arabidopsisthaliana)[11]、番茄(Lycopersiconesculentum)[12]和其他植物中均已进行研究报道。【本研究切入点】超敏反应是植物防御反应中细胞程序性死亡的一类。在灰霉菌侵入后,小立碗藓会激发与类似超敏反应(Hypersensitive response-like,HR-like)的防卫反应,在实验室(苔藓分子生物学与运用研究室)前期,通过转录组测序结果,比较对照组和灰霉菌处理12 h、1 d、2 d和3 d后小立碗藓的转录组数据,得到23个参与诱导细胞程序性死亡相关差异表达基因。其中PpCASPLUU3基因在灰霉菌侵染后的12 h与其他基因相比,该基因的表达量变化最为明显,表明PpCASPLUU3基因可能参与了由灰霉菌引起的小立碗藓类过敏反应。而PpCASPLUU3(GenBank:XM 024524752;类半胱氨酸蛋白酶)存在于小立碗藓中[13]。对苔藓植物中PpCASLUU3基因的研究尚未见报道。【拟解决的关键问题】构建小立碗藓PpCASLUU3的敲除载体,为后续研究PpCASLUU3是否参与了灰霉菌引起的小立碗藓中类过敏反应,对早期登陆植物的防卫防御的进化提供基础。
1 材料与方法
1.1 材料
1.1.1 试验材料 小立碗藓和质粒pTN182(5006 bp,用于构建PpCASLUU3基因敲除载体)由首都师范大学何奕騉教授提供,PTN182质粒中含有nptII,该基因在大肠杆菌中表现为抗卡那霉素。E.coliDH5α(用于外源基因的转化)由贵阳医学院提供。E.coliDH5α感受态细胞由贵州师范大学苔藓分子生物学与运用研究室制备。
1.1.2 试验仪器 冷冻离心机(湖南湘仪实验室仪器有限公司,TGL-16K),PCR仪(美国SCILOGEX公司,TC1000-G),恒温摇床培养箱(上海博迅实业有限公司,THZ-92A),超净工作台(苏州净化设备公司,SW-CJ-2D),凝胶成像系统(美国Bio-Rad公司,76S /07904),恒温水浴锅(上海博迅实业有限公司,HHS),电子天平(美国赛多利斯,TP-214),电泳仪(美国 Bio-Rad公司,A101439),灭菌锅(上海博迅实业有限公司,YXQ-LS-75)。
1.1.3 主要试剂 DNA提取试剂盒、胶回收试剂盒、EcoR Ⅰ、EcoR Ⅴ、BamH Ⅰ、SmaⅠPremixTaqTM、T4-DNA连接酶、氨苄青霉素(Amp)、1.5K DNA Marker均购于Takara。Plas-mid Mini Kit I试剂盒购于OMEGA biotek,卡那霉素购于美国sigma公司,酵母提取物(yeast ex-tract)和胰蛋白胨(tryptone)购于英国OXOID公司,葡萄糖、氯化钠、氢氧化钠及琼脂等试剂均购于国药集团化学试剂有限公司。引物的合成和序列测定工作,均委托武汉金开瑞生物工程有限公司完成。
1.2 方法
1.2.1 酶切位点的选择 在NCBI上检索(Genebank:XM_024524752.1)得到PpCASLUU3的DNA序列,将PpCASLUU3的DNA序列1414 bp分成上游臂(PpCASLUU3.1、700 bp)和下游臂(PpCASLUU3.2、714 bp)2段,用DNAMAN软件进行酶切位点分析,结合PTN182载体序列,最终选取EcoR I、EcoR V作为构建载体上游臂的酶切位点,SmaI、BamH I作为下游臂的酶切位点。利用Primer Primer 5软件设计扩增上、下游目的片段的引物(表1),在引物前端加入酶切位点序列,根据GC含量添加合适的保护碱基[19]。然后送武汉金开瑞生物工程公司合成,作为获取上游(PCR产物大小为522 bp)、下游(PCR产物大小为548 bp)目的片段的PCR扩增引物。
1.2.2 目的基因的获取 取培养20 d的小立碗藓新鲜材料,超纯水洗净,用液氮充分研磨至粉末,参照DNA提取试剂盒说明书提取DNA,以提取的小立碗藓DNA为模板,PpCASLUU3.1-F、PpCASLUU3.1-R为上游臂引物扩增上游臂片段,同样,以PpCASLUU3.2-F、PpCASLUU3.2-R为下游臂引物扩增下游臂片段。PCR反应体系如下表2,反应条件:94 ℃预变性5 min,94 ℃变性30 s,58 ℃退火30 s,72 ℃延伸1 min,35个循环,72 ℃再延伸10 min,4 ℃保存。将PCR后的产物进行1 %的琼脂糖凝胶电泳检测。检测后根据胶回收试剂盒说明书对目的片段进行割胶回收,获得目的基因。
1.2.3 PTN182质粒提取 取含PTN182质粒的大肠杆菌加入含卡那霉素的LB液体培养基中37 ℃ 220 r/min过夜培养,取5 mL培养液,根据Plas-mid Mini Kit I试剂盒说明书抽提PTN182质粒。

表1 PpCASLUU3上下游片段扩增的引物序列Table 1 Primer sequences of PpCASLUU3 upstream and downstream segments amplification
注:表中划横线部分为酶切位点。
Note: The underlined part in the table was the enzyme cutting site.

表2 PpCASLUU3上下游片段扩增的PCR反应体系Table 2 PCR reaction system of PpCASLUU3 upstream and downstream fragment amplification
1.2.4 PpCASLUU3.1-PMD19-T、PpCASLUU3.2-PMD19-T载体构建 将上述割胶回收的目的基因按表3的连接体系,16 ℃连接30 min,采用CaCl2法制作大肠杆菌感受态细胞[20]。将连接液加入到100 μl的大肠杆菌感受态细胞中,轻摇混匀,冰浴30 min,42 ℃热激90 s后立即冰浴3 min,加入900 μl不含氨苄青霉素(Amp)的LB液体培养基,37 ℃震荡培养1 h,12 000 r/min离心1 min,取100 μl上清液重悬沉淀,将重悬的菌液涂布于含Amp抗性的LB平板,放置数分钟,待菌液干后放入37 ℃恒温培养箱内倒置培养16 h。待其长出单菌后,挑取单菌于含Amp的LB液体培养基中37 ℃恒温摇床培养箱220 r/min培养16 h,取2 μl菌液,以表1中的引物做菌液PCR,筛选能扩增出目的片段的菌液,用Plas-mid Mini Kit I试剂盒进行质粒抽提。得到PpCASLUU3.1-pMD19-T质粒,用EcoR I、EcoR V酶切验证。使用相同的方法得到PpCASLUU3.2-pMD19-T质粒。
1.2.5 PpCASLUU3.1-PTN182载体构建 使用EcoR I、EcoR V同步酶切PpCASLUU3.1-pMD19-T和PTN182原始载体,酶切体系如表4所示,37 ℃酶切3 h后加入5 μl loading buffer进行1 %的琼脂糖凝胶电泳,割取目的片段大小的条带及酶切后的PTN182载体回收,然后按表4连接体系进行16 ℃连接16 h,取连接产物5 μl转入大肠杆菌感受态细胞,转化方法同1.2.2。取100 μl转化液涂布于含卡那霉素的LB平板,待菌液干后置于37 ℃恒温培养箱内倒置培养16 h。挑取单菌于含卡那霉素的LB液体培养基中37 ℃恒温摇床培养箱220 r/min培养16 h,后取2 μl菌液做菌液PCR筛选,将能扩增出目的片段的菌液进行质粒抽提,酶切验证,选取阳性质粒送武汉金开瑞生物工程公司测序。

表3 PpCASLUU3.1-pMD19-T及PpCASLUU3.2-pMD19-T载连接体系Table 3 PpCASLUU3.1-pMD19-T and PpCASLUU3.2-pMD19-T vector connection system
1.2.6 PpCASLUU3.1-PTN182-PpCASLUU3.2载体构建 将含有PpCASLUU3.2片段的 PpCASLUU3.2-pMD19-T质粒和PpCASLUU3.1-PTN182质粒分别摇菌过夜,取5 mL培养液进行质粒抽提,PpCASLUU3.2-pMD19-T用SmaI和BamH I进行双酶切,回收切出片段,与同样酶切的PpCASLUU3.1-PTN182载体连接,获得重组质粒PpCASLUU3.1-PTN182-PpCASLUU3.2(图1),具体方法同1.2.3。
2 结果与分析
2.1 PpCASLUU3.1、PpCASLUU3.2目的基因的获取
通过设计引物扩增PpCASLUU3.1、PpCASLUU3.2目的基因,将小立碗藓总DNA经过PCR反应后用琼脂糖凝胶电泳检测(图2)可见:A凝胶图上1号泳道中的PCR产物的条带与PpCASLUU3.1目的基因大小条带相同(500 bp左右),B凝胶图上的1号泳道中的PCR产物的条带与PpCASLUU3.2目的基因条带大小相同(500 bp左右),A、B图中阴性对照在500 bp左右的位置未出现条带。表明该引物设计合理,能正常扩增出目的片段,可用于目的基因的获取。
2.2 PpCASLUU3.1-pMD19-T及PpCASLUU3.2-pMD19-T载体构建
如图3所示,A中1号泳道为PpCASLUU3.1-pMD19-T质粒用EcoR I、EcoR V酶切后的琼脂糖凝胶电泳图,B中1号为PpCASLUU3.2-pMD19-T质粒用BamH I、SmaI酶切后的琼脂糖凝胶电泳图,2号均为未经过酶切的原始质粒,两者质粒酶切后在泳道的相应位置均出现了与目的片段(500 bp左右)大小一致的条带。说明,PpCASLUU3.1、PpCASLUU3.2片段成功的克隆到pMD19-T载体中。
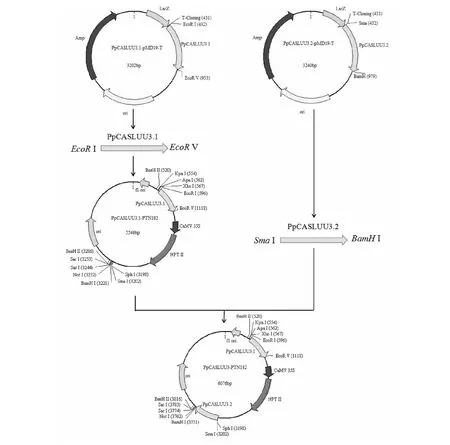
图1 PpCASLUU3-PTN182载体构建示意图Fig.1 Schematic diagram of PpCASLUU3-PTN182 vector construction

表4 目的片段及载体体系Table 4 Objective fragment and vector enzyme cutting system

A图中1为PpCASLUU3.1目的基因,2为阴性对照。 B图中1为PpCASLUU3.2目的基因,2为阴性对照。 M:Marker= 15 000 bpFig.A1, PpCASLUU3.1; 2. Negative control. Fig.B 1, PpCASLUU3.2; 2. Negative control. M: Marker= 15 000 bp图2 PpCASLUU3.1、PpCASLUU3.2目的基因的获取Fig.2 Acquisition of target genes of PpCASLUU3.1 and PpCASLUU3.2
2.3 PpCASLUU3.1-PTN182-PpCASLUU3.2载体构建
通过筛选后的质粒用EcoR I、EcoR V酶切后如上图4A中的1号泳道所示,用BamH I、SmaI酶切验证如上图4B中的1号泳道所示,两者均能切出与目的片段大小的条带,2号为对照(未用限制性内切酶酶切),在目的片段大小的位置处无条带出现,表明PpCASLUU3.1、PpCASLUU3.2目的片段均已插入PTN182中。
由图5可见,经测序PpCASLUU3.1-PTN182-PpCASLUU3.2 载体的PpCASLUU3.1序列与原始PpCASLUU3.1序列比对,相似高度达100 %。而载体上的PpCASLUU3.2序列与原始PpCASLUU3.2序列比对,相似度为99.63 %,证明PpCASLUU3.1-PTN182-PpCASLUU3.2敲除载体构建成功。

A为PpCASLUU3.1-pMD19-T质粒酶切验证图,1表示用EcoR I、EcoR V酶切的质粒,2表示未酶切的PpCASLUU3.1-pMD19-T质粒。B为PpCASLUU3.2-pMD19-T质粒酶切验证图,1表示用BamH I、Sma I酶切的质粒,2表示未酶切的PpCASLUU3.2-pMD19-T质粒,M:Marker = 15 000 bpNote: A. Verification diagram of plasmid digestion of PpCASLUU3.1-pMD19-T. 1, plasmid digestion with EcoR I and EcoR V; 2, plasmid digestion of PpCASLUU3.1-pMD19-T without enzyme digestion. B, enzyme digestion verification diagram of PpCASLUU3.2-pMD19-T plasmid, 1, plasmid digested by BamH I and Sma I; 2, plasmid digested by PpCASLUU3.2-pMD19-T without enzyme digestion, M: Marker = 15 000bp图3 PpCASLUU3.1-pMD19-T、PpCASLUU3.2-pMD19-T质粒酶切验证图谱Fig.3 PpCASLUU3.1-pMD19-T and PpCASLUU3.2-pMD19-T plasmid digestion verification diagram

A 图中1用EcoR I、EcoR V酶切的质粒,2未经过酶切的PpCASLUU3.1-PTN182-PpCASLUU3.2质粒。B图中1用BamH I、Sma I酶切的质粒,2表示未经过酶切的PpCASLUU3.1-PTN182-PpCASLUU3.2质粒A, plasmid 1 was digested by EcoR I and EcoR V; 2, undigested PpCASLUU3.1-PTN182-PpCASLUU3.2 plasmid. B, plasmid 1 was digested by BamH I and Sma I; plasmid 2 was undigested PpCASLUU3.1-PTN182-PpCASLUU3.2图4 PpCASLUU3.1-PTN182-PpCASLUU3.2质粒酶切验证图谱Fig.4 PpCASLUU3.1-PTN182-PpCASLUU3.2 plasmid digestion verification diagram

图5 PpCASLUU3.1与PpCASLUU3.2插入片段测序Fig.5 PpCASLUU3.1 and PpCASLUU3.2 insert fragment sequencing results
3 讨 论
随着生物技术的发展,构建敲除载体的方法也越来越多,如Gateway、in-fusion、TA克隆技术等。Gateway技术是一种快速构建载体的方法,主要利用噬菌体体外位点特异性重组构建载体,其特点是反应时间短,不需要限制性内切酶和连接酶参与便可完成[21]。in-fusion是一种快速将片段与载体进行无缝连接的方法,能够一次性插入多个片段,无需酶切和连接步骤,操作简单,转化率高等特点[22]。但以上两种方法成本都较高[21]。而TA克隆技术(TA cloning)是利用Taq聚合酶具有末端转移酶(TdT)活性,但却不具有3′和5′端外切酶校准活性的特点,可在PCR产物的3′端加上一个非模板依赖碱基A。pMD19-T载体的3′携带一个碱基T。能高效地与带A尾的PCR产物连接,极大地提高了克隆的效率。TA克隆是目前克隆PCR产物最简便、快捷的方法[23]。鉴于此,试验选用了TA 克隆技术将PCR产物直接克隆到pMD19-T中,测序验证后将其酶切、连接到PTN182载体中,此法的酶切效率比直接酶切PCR产物高,可获得较纯的酶切片段,使得后续实验更为顺利。在获取PCR产物时,以小立碗藓DNA为模板,使用引物设计软件设计的小立碗藓PpCASLUU3基因上、下游同源臂的引物,利用PCR技术扩增得到小立碗藓PpCASLUU3基因的上、下游同源臂,如图2A、B中的2号泳道(对照)出现的条带均低于250 bp,这可能是引物自身结合形成的二聚体所导致,试验采取割胶的方法回收PCR产物,此结果对后续试验无影响。
在载体构建过程中,酶切位点的选择尤为关键,试验在同一载体中分2次插入,进行酶切位点分析的时候应将2条片段同时放入DNAMAN软件中进行分析,避免所选择的酶在另一片段中存在作用位点。尽量选择一些能共用Buffer、作用温度相同的酶,这样可以进行同步酶切,相对于分步酶切可节约大量时间。限制行内切酶在识辨太近、以及共用碱基对的酶切位点时会出现错误,所以在选择酶切位点时应尽量避开两个酶切位点之间碱基相隔少、共用碱基对这两者因素,因此在选择载体的酶切位点时需避开以上述情况,保证载体能够充分的被切开。
构建载体时若采用内切酶直接切割PCR产物的方式进行载体构建,得到的阳性克隆极少,载体构建效率低下。而采用此法后,得到的阳性克隆明显增多,载体构建的效率得到极大的提升。
4 结 论
用T4-DNA连接酶将酶切得到的目的片段与相同酶切后的PTN182载体连接,通过PCR阳性检测、酶切和测序验证等结果证明PpCASLUU3.1-PTN182-PpCASLUU3.2敲除载体构建成功。该载体的构建成功,为小立碗藓中PpCASLUU3基因敲除提供了条件,为PpCASLUU3基因功能的研究奠定了基础。
猜你喜欢
中国土壤与肥料(2021年5期)2021-12-02 01:05:52
现代仪器与医疗(2021年2期)2021-07-21 02:19:16
中国当代医药(2020年26期)2020-11-06 07:22:10
疯狂英语·新悦读(2020年7期)2020-07-30 01:32:52
生物信息学(2020年2期)2020-07-09 01:27:56
癌变·畸变·突变(2020年1期)2020-02-12 05:18:04
系统工程与电子技术(2016年2期)2016-04-16 05:16:53
上海蔬菜(2015年2期)2015-12-26 05:03:40
分析化学(2015年10期)2015-11-03 07:38:01
中国光学(2015年1期)2015-06-06 18:30:20
