
大豆抗菌核病的全基因组关联研究
2020-04-16 03:41FranoisBelzile
华北农学报 2020年1期
张 羽,François Belzile
(1.陕西理工大学 生物科学与工程学院,陕西 汉中 723000;2.Département de Phytologie,Université Laval,Québec,QC,Canada G1V 0A6)
大豆菌核病是20世纪80年代中期开始出现的世界范围大豆病害,主要分布在美国、加拿大、中国、阿根廷、匈牙利、日本和印度等国,特别是在北美和加拿大的寒凉大豆种植区已经成为影响大豆产量的主要病虫害之一[1-2]。它主要由核盘菌(Sclerotiniasclerotiorum(Lib.)de Bary)侵染寄主植株所引起。1986年,加拿大的豆类作物菌核病发病率达25%[3]。近年来,大豆菌核病有继续扩大蔓延、加重危害的趋势,严重威胁大豆的生产[4]。生产实践证明,培育抗(耐)病大豆品种是防治大豆菌核病最经济有效的途径。目前为止,还没有发现完全免疫菌核病的大豆资源,前人研究报道,大豆种质资源对菌核病的抗性存在着遗传差异,有的大豆资源具有部分抗性,大豆菌核病是由多基因控制的数量性状遗传。数量性状对环境条件敏感,在大田条件下目标性状的选择压力难以保证一致,导致筛选抗菌核病种质资源比较困难,因此,利用分子标记辅助选择培育抗性品种显得越来越重要。
大豆抗菌核病的QTLs定位自2000年Kim等[5]利用分子标记RFLP、SSR和RAPD进行研究以来,很多研究都是基于SSR分子标记对大豆抗菌核病进行QTLs定位,但由于一代和二代分子标记在基因组中的多态性有限,使其在QTLs定位中的利用不太理想,随着二代测序技术的发展,基于SNPs的全基因组关联研究在生物领域成为热点,自从全基因组关联研究在人类疾病研究中取得进展后,植物上通过全基因组关联研究进行病害候选基因定位也越来越受到重视。由于全基因组测序面临费用高和较多重复序列的缘故,为了提高测序性价比,基于限制性核酸内切酶识别位点相关DNA序列(RAD tags)的简化基因组测序技术得到了普遍应用,其中GBS(Genotyping by sequencing)技术由于能大幅降低基因组的复杂度,尤其适合于大样本量的研究,可快速鉴定出高密度的SNPs位点,其结果在QTLs定位中应用越来越广[6-12]。目前为止,在大豆上多选择Apek Ⅰ、MspⅠ和PstⅠ酶进行简化基因组后测序,基于SNPs的全基因组关联研究进行大豆菌核病QTLs定位的研究报道较少,由于定位样本量大小、表型鉴定方法、分析算法及标记多态性位点数目等因素,影响了QTLs定位的一致性。且由于定位材料及标记数目不同,研究结果也不一致[13-16]。分子标记辅助选择培育大豆抗菌核病育种还未见报道,抗性种质资源的选择很多还是用草酸浸泡叶、根、茎后,根据植株萎蔫程度和茎中可溶性色素水平高低评价抗性强弱[17],因此,大豆菌核病QTLs的定位对大豆抗性育种及抗性机理的研究进展相对缓慢。本研究通过对126个加拿大大豆品种的30 125个SNPs,利用PLINKv 1.07分析软件,进行SNP-phenotype和Haplotype-phenotype的关联分析,找到大豆与抗菌核病强关联的位点/候选基因,为大豆菌核病QTLs分析及大豆抗菌核病育种提供参考。
1 材料和方法
1.1 DNA提取、文库准备、测序、数据质量控制
126个大豆品种的DNA提取、文库准备、测序、质量控制等步骤参考文献[13]。
1.2 分析方法
1.2.1 表型值 用棉垫接种法接种核盘菌菌丝进行表型鉴定[18],茎秆损伤长度介于28.6~192.4 mm,供试材料的表型值分布见图1。
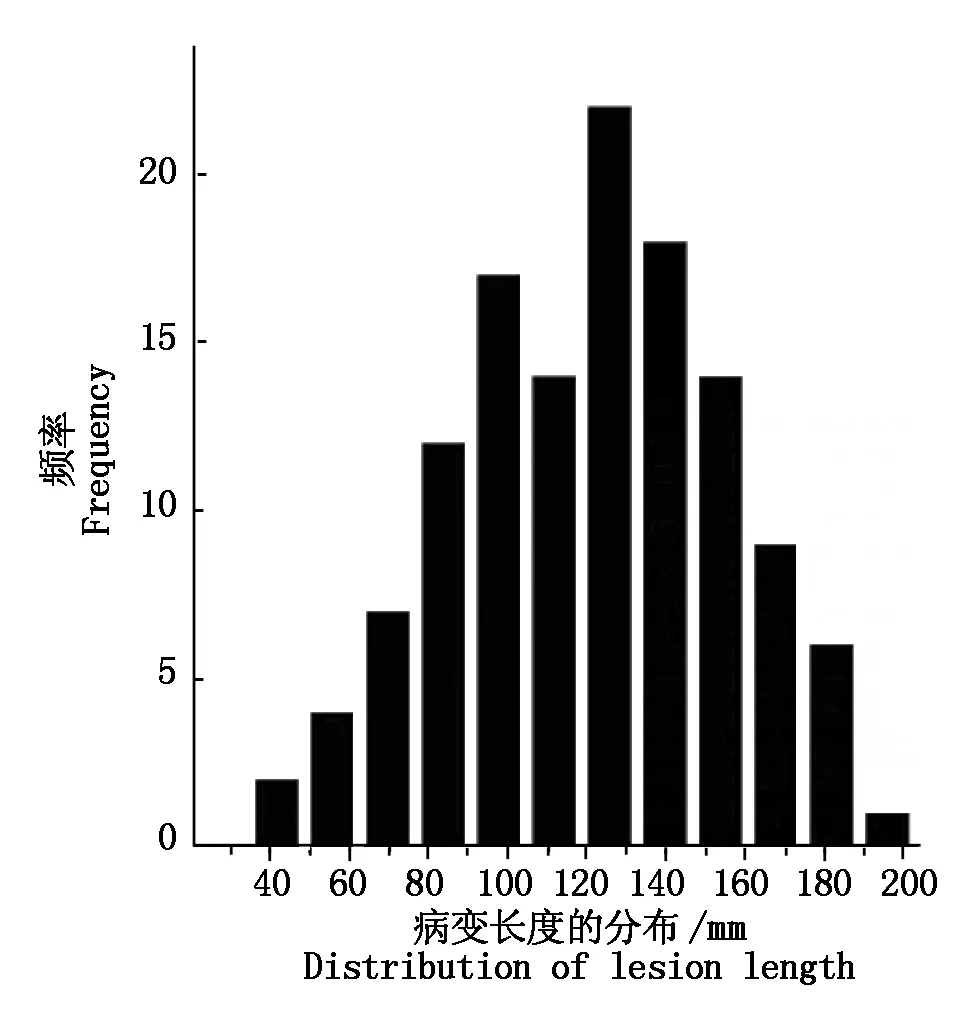
图1 供试材料主茎损伤长度表型值分布
1.2.2 数据质量控制与分析 数据质量控制是全基因组关联分析的关键之一,不同参数的选择代表了不同的遗传学意义,产生不同的分析结果,不当的参数选择会产生假阳性或假阴性结果。本研究位点间的关联程度r2值0.8,LOD取值3,置信区间为95%,MAF值取0.01。由于大豆为自交作物,偏离哈迪-温伯格平衡的SNPs很可能是与病害关联的易感位点,因此,本研究不考虑哈迪-温伯格平衡,得到30 125个SNPs(图2)。126个材料共有3.7958E6个位点,其中杂合基因型位点有248 456个,占6.546%,杂合体的质量控制在10%以下,说明了数据可信度高[19-21]。
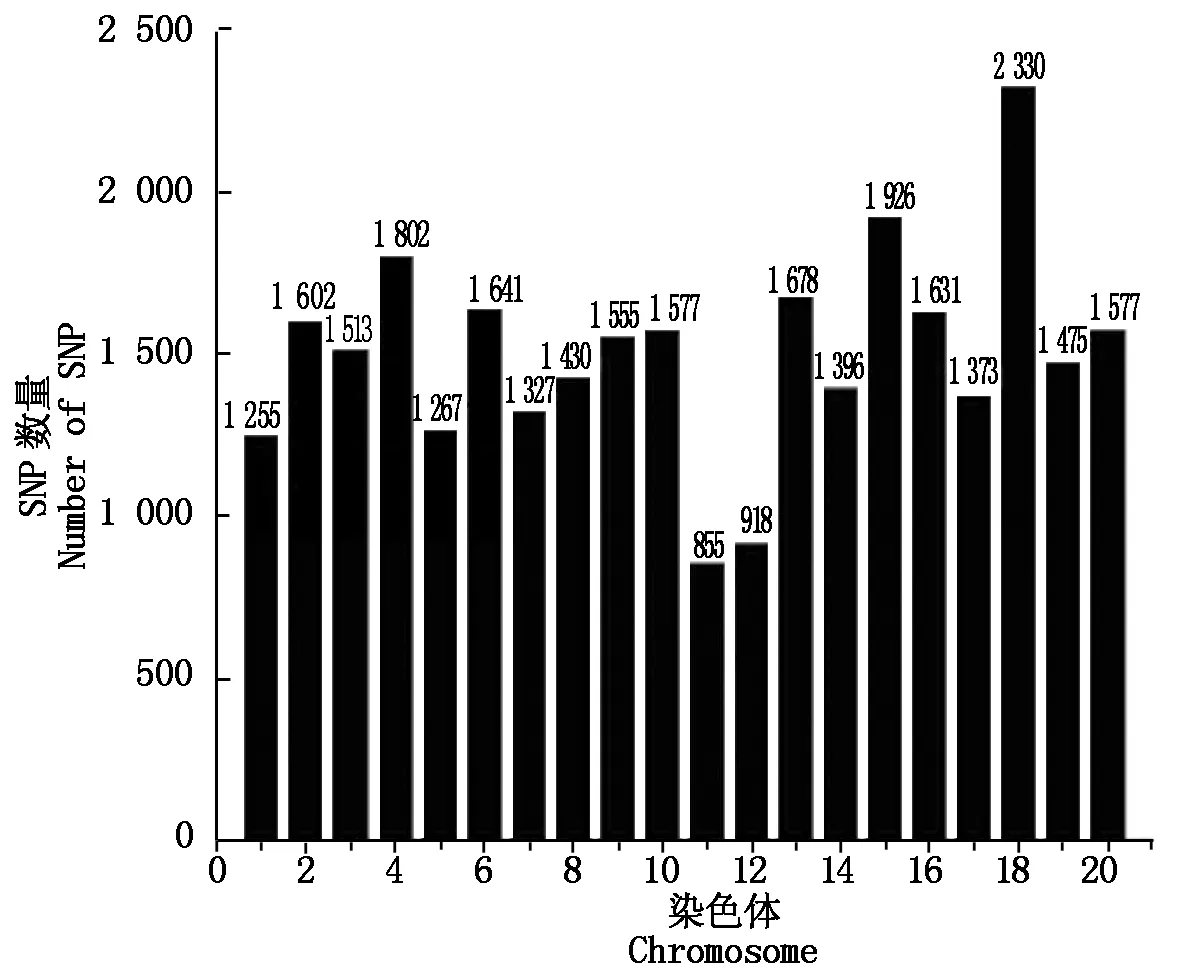
图2 每条染色体上的SNP数
2 结果与分析
2.1 126份大豆材料的NJ法聚类分析
用TASSEL 5.0的邻接法(The neighbor-joining algorithm,NJ)对126个供试材料进行聚类(图3),聚为3大类,分别为67,45,14个材料。
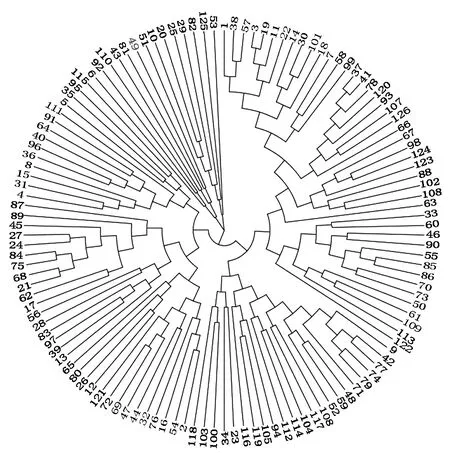
图3 基于NJ法的聚类图
2.2 PC分析
用SPSS对126份大豆品种进行主成分分析,结果显示前2个主成分的方差贡献率分别为6.24%和5.44%,Ward法聚类结果见图4。126份供试材料聚为2类,分别为70,56个材料。
2.3 群体结构分析
很多研究表明,群体结构分析是对目标性状进行关联分析的前提,因为复杂的群体结构会导致基因型与表型之间的假阳性。研究发现,考虑群体结构和不考虑群体结构时检测到的与表型变异相关的多态性位点数不同[22]。本研究采用Structure 2.3.4软件模拟群体遗传结构[23],其基本原理是:假设样本存在K个(K值未知)等位变异频率特征类型数,将供试材料归到K个群体,使得该群体位点频率都遵循同一个Hardy-Weinberg 平衡。设定群体数K的取值为1~10,将MCMC(Markov chain Monte Carlo)开始时的不作数迭代(Length of burn-in period)设置为10 000 次,再将不作数迭代后的MCMC设置为10 000 次,每个K运行20次,然后依据ΔK值最大的原则选取一个合适的K 值。本研究的ΔK值最大时的K值为2,见表1。将126个供试材料划分为2个亚群,分别为68,58个材料。

图4 Ward法聚类图


表1 群体结构分析
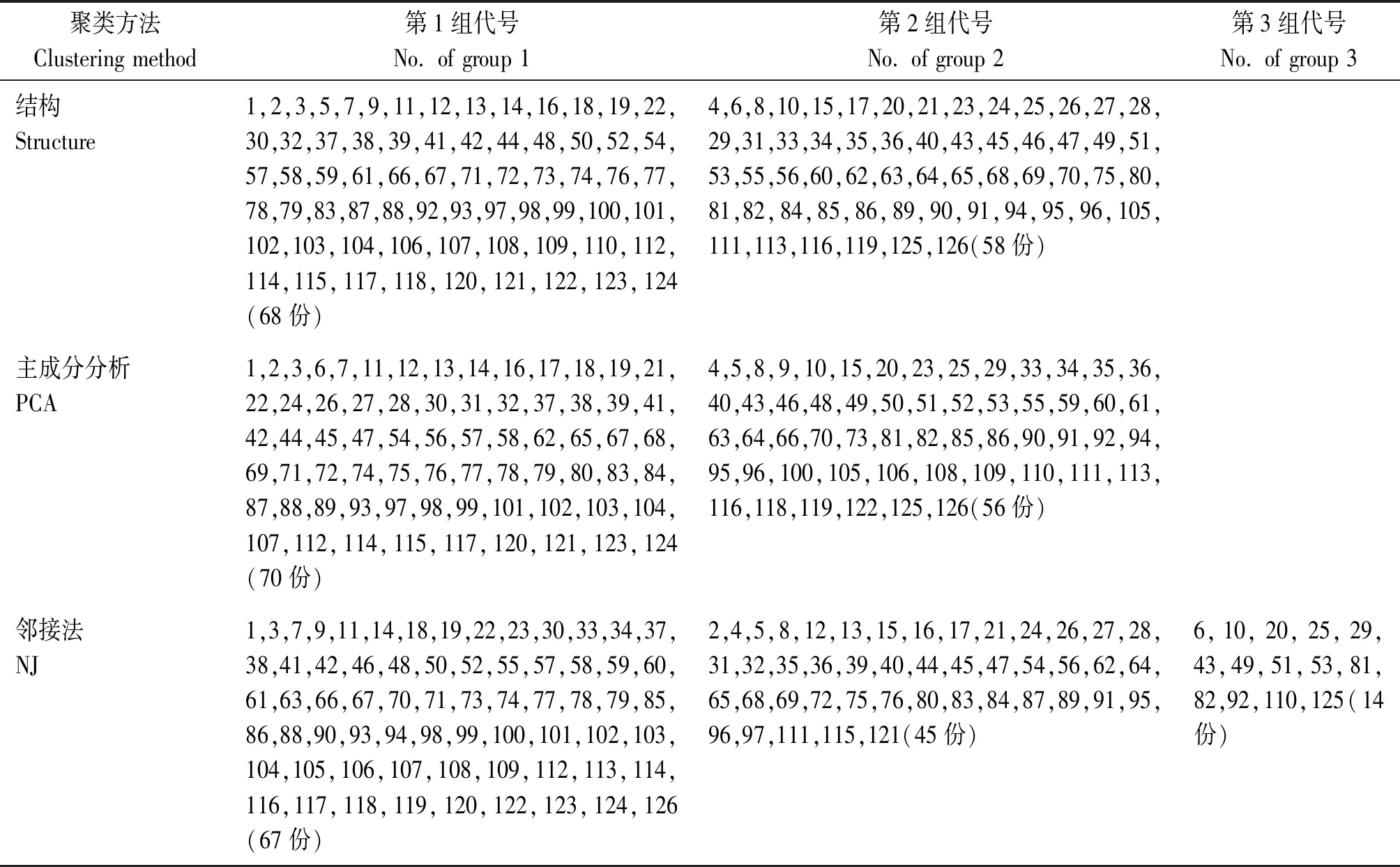
表2 用Structure、PCA 和UPGME方法的聚类结果比较
2.4 连锁不平衡分析
用PLINKv 1.07命令“--r2”运行LD,过滤掉r2小于0.2的数据,共有82 911对,r2平均值为r0.73,r2大于等于0.8的有46 655对(占56.27%),2等于1的有9 391对(占11.33%)。2个SNPs位点间相关(r2大于0.2)的平均距离为114 979.2 bp,r2小于0.8的平均距离为124 596.9 bp,r2大于等于0.8的平均距离为107 505.3 bp,r2等于1的平均距离为80 397.08 bp。两位点间距离≤500 kb(遗传学上把位点间大于500 kb认为没有连锁关系)的r2平均值为0.73。由此看出,LD随着2个位点间的距离增大而衰减。从单条染色体和全基因组r2和2个SNPs位点间距离的关系图也可以看出随着物理距离的增加,2个位点间的r2越小,即连锁强度越小,甚至没有(图5,6)。
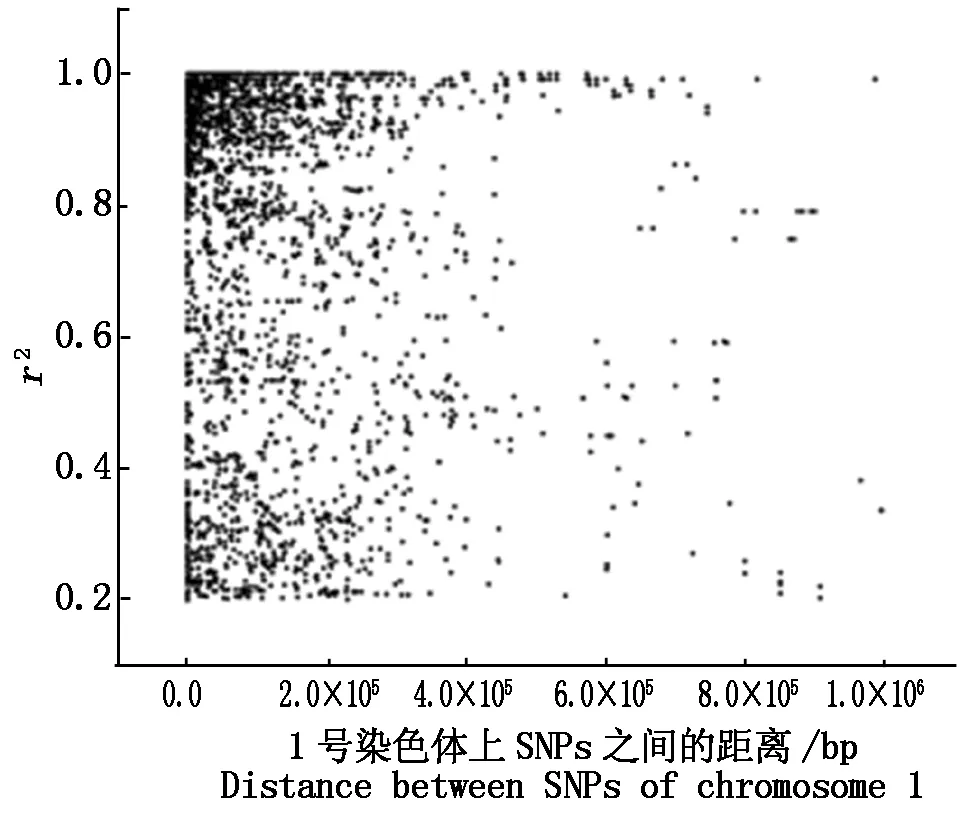
图5 一号染色体上的LD衰减图
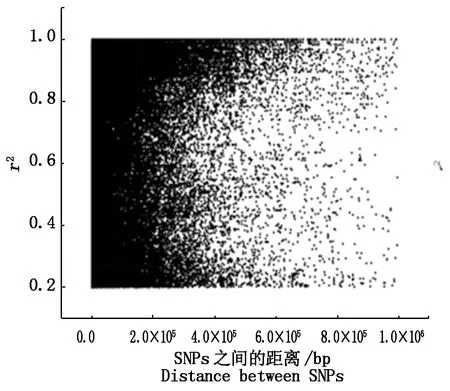
图6 全基因组上的LD衰减图
2.5 单体型分析
SNPs位点并不是独立遗传的,而是在染色体上倾向于以一个整体遗传给后代,成组遗传的SNPs位点在一代又一代的遗传中绝少发生重组,于是,这样的一组SNPs位点类型也就是单体型,也就是说相邻位点是以单体型为单位传递的(前提是位点间的LD很强),当然,单体型分析在统计学上的作用主要是减少多个位点组合分析时的自由度,从而增加研究的效能,例如,如果要分析3个二等位多态位点的组合和疾病/病害的关系,理论上有9种不同的组合,但是因为这3个位点间不是独立的,在群体中可能只存在3种常见的单体型,如果3个位点间独立,也就是连锁平衡,那么群体中的单体型就会有9种,这时候做单体型分析是没有意义的,因此,进行单体型分析时最好以单体域Block为基础。在PLINKv 1.07里有2种单体域推断方法,Sliding windows和Haplotype list,Haplotype list要先计算Blocks,然后利用算出的Haplotype和Phenotype之间的关联程度进行分析,这种运算比Sliding windows灵活,因为Sliding windows只能按SNP顺序依次排列分析Haplotype,不能按实际跳跃分析。在比较了2种方法后,本研究采用“--blocks”命令推断Blocks。20条染色体共有2 863个Haplotype,它们在20条染色体上的分布见图7,其中18号染色体上的Haplotype最多(230个),11号染色体上的最少(57个)。一个Haplotype最多的由36个SNPs组成,最少2个SNPs组成。最长Haplotype跨越200 kb(因此,后续候选基因在200 kb范围内注释),最短跨越2 bp。
2.6 基因型与表型关联分析
在基因型-表型的关联中,为了发现较多的关联信息,把P小于1E-5的分析结果都列表显示。
2.6.1 基于Haplotype-phenotype关联分析 基于Haplotype-phenotype关联分析中,17号染色体上的4个SNPs组成的Haplotype解释表型变异最大(17.56%)。其余依次为1,3,4,20号染色体上的Haplotype(表3)。

图7 每条染色体上的Haplotype数
2.6.2 基于单个SNP-phenotype关联分析 在单个SNP的基因型-表型关联分析中,α≤0.05(P≤0.05/30125=1.660E-6)时,最强关联在3号染色体的34387780位置和其临近的几个SNP位点(P值为8.669E-7),可解释表型变异的17.80%。在单个SNP-phenotype的关联分析中发现,往往多个SNPs组成了一个Haplotype,如在3号染色体上,从SNP3818到SNP3823一共6个SNP组成了一个Haplotype,这种情况同时也在其他染色体上看到。但由于126个材料中某些材料在一个Haplotype的几个SNPs中的个别SNP位点为杂合子,所以导致单个SNP解释的表型变异不一致(表4),但峰值SNP都在相应的Haplotype中。其次关联在20,1,4,17号染色体上(图8)。
2.7 候选基因
由于峰值SNP都出现在相应的Haplotype中,因此结合单个SNP和Haplotype的基因型-表型关联分析位点,本研究将较强关联的位点在Soybeanbase数据库(http://soybase.org/tools.php)中比对分析,可能参与大豆菌核病防御机制的蛋白质见表5,大致可分为参与生物过程、细胞组成、分子功能及未知功能蛋白。
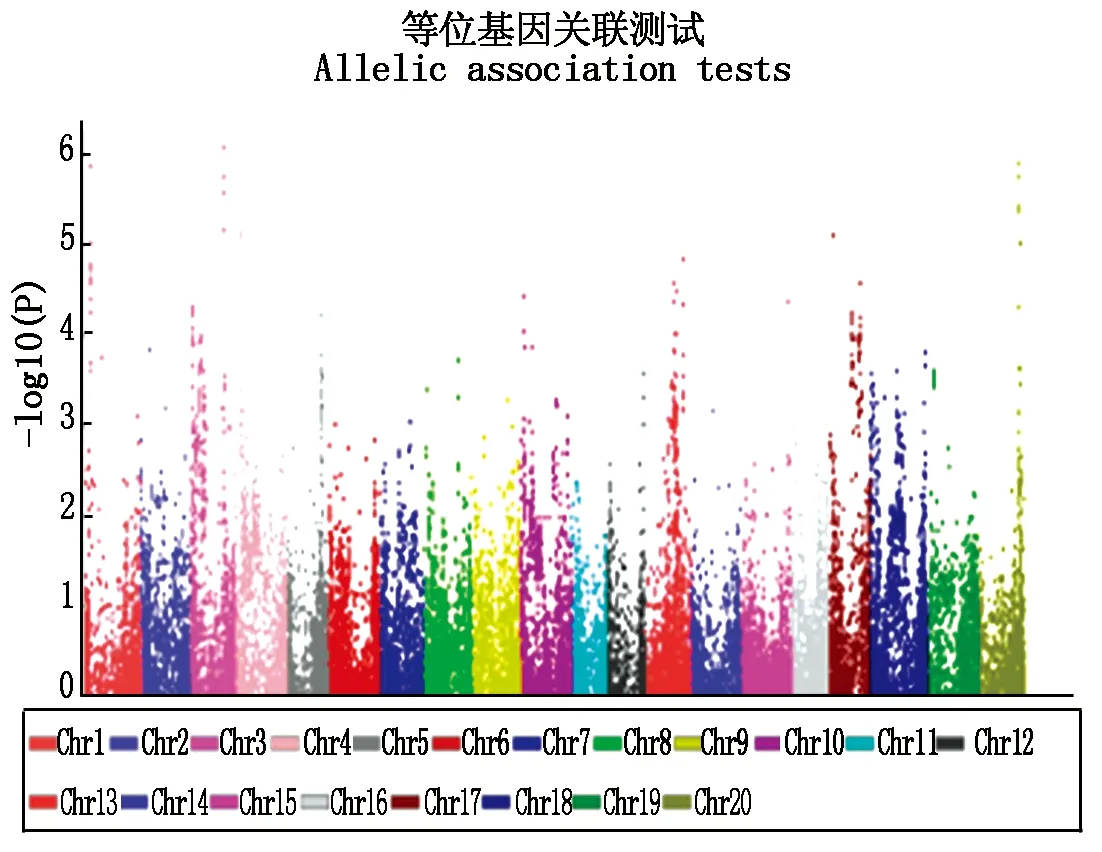
图8 基于单个SNP的基因型-表型关联

表3 用PLINYKv 1.07进行的基于Haplotype-表型关联分析

表4 用PLINYKv 1.07进行的基于单个SNP-表型关联分析
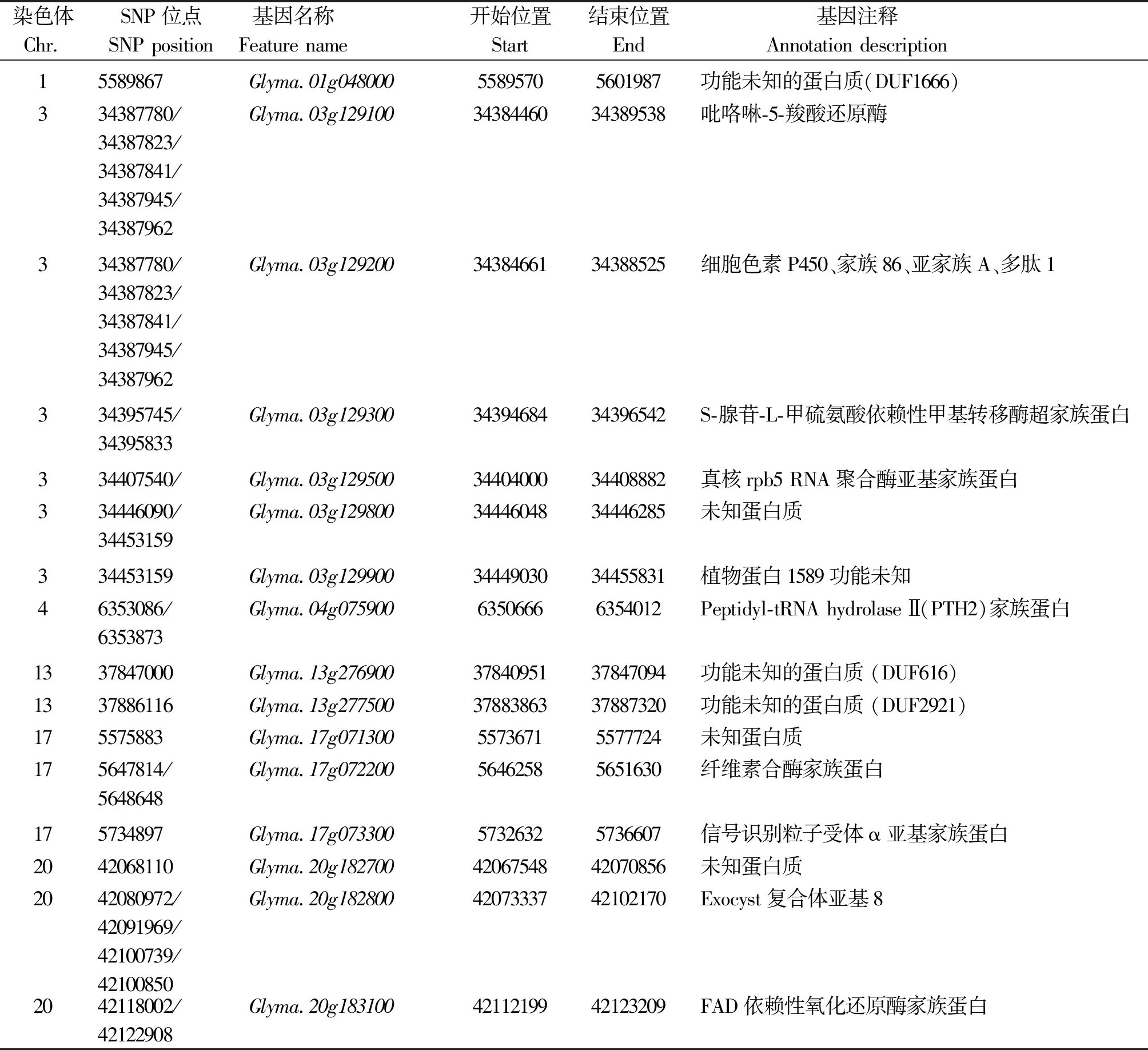
表5 候选基因
3 讨论
核盘菌侵染寄主的致病机理非常复杂,目前认为核盘菌分泌的细胞壁降解酶(Cell wall degrading enzymes)和草酸在致病中起重要作用,由于草酸氧化酶(Oxalate oxidase,OXO)和草酸脱羧酶(Oxalate decarboxylase,OXDC)能把草酸降解成CO2和H2O2,从而减少草酸的危害,降低了病原菌的侵染,研究表明把从小麦和大麦中分离得到的OXO(一种类萌芽素蛋白 germin-like proteins)和OXDC转化进大豆中能提高大豆对菌核病的抗性[25-27]。研究发现,核盘菌致病仅有细胞壁降解酶或草酸还是不能完全致病,需要其他因子协同才能具有致病力。大豆抗菌核病的许多研究都是通过鉴定接种菌丝体后,不同时间段大豆植株抗逆酶谱/防御相关基因的表达谱的变化和细胞壁胼胝质沉积来鉴定有哪些抗逆酶参与了此过程,前人用蛋白组学技术鉴定出超氧化物歧化酶(SOD)、多酚氧化酶(PPO)、过氧化物酶(POD)、苯丙氨酸解氨酶(PAL)等在菌核病侵染过程中抗感材料的活性有所变化[28]。本研究中没有关联出OXO、OXDC、SOD、PPO、POD、PAL等基因。
本研究基于Haplotype-Traits的最大关联在17号染色体上,在此区域内的纤维素合酶基因(Glyma.17g072200)被鉴定出关联。植物细胞壁主要由果胶质、纤维素、木质素等组成,是病原物侵入寄主植物的主要屏障。植物没有强大的免疫系统,所以自身的防御机制就显得尤为重要,细胞壁是植物抵御外来侵染的第一道屏障,在油菜中有研究报道,菌核病菌菌株的致病力与其纤维素酶活性呈正相关[29-30]。另外,Valera[31]从生理、解剖和分子技术对大豆菌核病的侵染过程进行了研究,感染不同阶段的大豆转录组测序研究证明感性品种的木质素生物合成与JA/ET-related defense responses关闭有关,证明了细胞壁特性与木质素酶在大豆抗菌核病中的重要作用。
基于单个SNP-Trait的最大关联在3号染色体上,在此范围内的候选基因有pyrroline-5-carboxylate(P5C)reductase(Glyma.03g129100)和cytochrome P450、family 86、subfamily A、polypeptide 1(Glyma.03g129200)。P5CR是广泛存在于原核和真核生物中的一种重要的管家蛋白,其主要功能是催化脯氨酸生物合成的最后一步反应,即在NAD(P)H的作用下,将吡咯啉-5-羧酸(P5C)转化为脯氨酸的反应。许多研究报道,在应激引起的损害中,脯氨酸含量在植物体内聚集[32-33]。植物中P450在寄主-病原物互作中的表达已经有较多证据[34-35]。
用全基因组SNP关联研究进行大豆菌核病QTLs定位显示出诸多优点,但QTLs定位工具的选择是QTLs定位研究中需要考虑的主要问题之一,在本研究中利用几种模型分析后发现,并不能断言某个软件或某种方法最适合QTLs定位,因此,在关联分析中尽可能多用几种软件或多种方法分析比较,然后对共同定位的位点进行进一步研究验证。
在与复杂性状的关联分析中,样本数和标记数多少影响定位效果,从理论上讲样本数目越多且标记覆盖基因组范围越大,其分析结果越客观。
本研究引入Haplotype-phenotype的关联可能提高真正与病害相关位点的检出率,从Haplotype关联的基因可以看出,与病害相关基因簇都包含在一个关联的Haplotype中,这些基因簇在病害与寄主应答中可能协同作用。通过对关联出的基因簇的分析,可以加深对病害应答通路的认识,Haplotype很可能作为一个功能单位在起作用。另外,在单个SNP和Haplotype与表型的关联分析中,峰值SNP都包含在其相应的Haplotype中,但由于Haplotype包含许多SNPs的遗传信息,采用单体型比单个SNP具有更好的统计分析效果,能降低自由度。
以α≤0.05为阈值,3号染色体上的候选基因为Glma.03g129100、Glma.03g129200、Glma.03g129300、Glma.03g129500、Glma.03g129800、Glma.03g129900。17号染色体上为Glma.17g071300、Glma.17g072200、Glma.17g073300。目前,通过关联分析在大豆的抗菌核病中也已经定位了一些候选基因,如何在抗病育种中有效应用这些QTLs,需要在分析的基础上进一步结合蛋白组学进行功能验证,直到成为稳定的标记才能在抗病育种中应用。
猜你喜欢
四川农业科技(2022年2期)2022-11-21
中国现代医生(2022年21期)2022-08-22
四川农业科技(2021年7期)2021-08-20
四川蚕业(2021年3期)2021-02-12
天津医科大学学报(2021年1期)2021-01-26
医药前沿(2020年20期)2020-11-10
三农资讯半月报(2020年2期)2020-03-09
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
中学生理科应试(2016年4期)2016-11-19
