
Congenital nephrogenic diabetes insipidus due to the mutation in AVPR2 (c.541C>T) in a neonate: A case report
2020-04-07 02:13:24FaTaoLinJingLiBangLiXuXiuXiuYangFangWang
World Journal of Clinical Cases 2020年24期
Fa-Tao Lin, Jing Li, Bang-Li Xu, Xiu-Xiu Yang, Fang Wang
Fa-Tao Lin, Jing Li, Bang-Li Xu, Xiu-Xiu Yang, Department of Neonatology, Children’s Hospital Affiliated to Zhengzhou University, Henan Children’s Hospital, Zhengzhou Children’s Hospital, Zhengzhou 450000, Henan Province, China
Fang Wang, Department of Infectious Diseases, Children’s Hospital Affiliated to Zhengzhou University, Henan Children’s Hospital, Zhengzhou Children’s Hospital, Zhengzhou 450000,Henan Province, China
Abstract BACKGROUND Congenital nephrogenic diabetes insipidus (CNDI) is a rare hereditary renal disorder that is caused by mutations in AVPR2 or aquaporin 2 (AQP2). Up to now, there are few reports about CNDI in neonates. Early clinical manifestations of CNDI in neonates are atypical. A lack of understanding of the disease by clinicians causes frequent misdiagnoses or missed diagnoses, which may result in failure to administer treatments in time and ultimately leads to severe complications. In this study, clinical data of a case of AVPR2 gene mutationinduced CNDI, which was confirmed by genetic testing, were retrospectively analyzed to improve our understanding of this disease.CASE SUMMARY On February 1, 2020, a male neonate was hospitalized 17 d after birth due to a 7 d period of pyrexia. The patient’s symptoms included recurrent pyrexia,hypernatremia and hyperchloremia, which were difficult to treat. The patient was fed on demand, and water was additionally provided between milk intakes. A combination treatment of hydrochlorothiazide and amiloride was administered.After the treatment, body temperature and electrolyte levels returned to normal,the volume of urine was significantly reduced and the patient was subsequently discharged. Genetic tests confirmed that the patient carried the AVPR2 gene missense mutation c.541C>T (P.R181C), and the patient’s mother carried a heterozygous mutation at the same locus. After clinical treatment with a combination of hydrochlorothiazide and amiloride, the body temperature and electrolyte levels returned to normal. Up until the most recent follow-up examination, normal body temperature, electrolyte levels and growth and development were observed.CONCLUSION CNDI in the neonatal period is rare, and its clinical manifestations are unspecific with some patients merely showing recurrent fever and electrolyte disturbance.Genetic testing of AVPR2 and AQP2 can be used for screening and genetic diagnosis of CNDI.
Key Words: Neonate; Congenital nephrogenic diabetes insipidus; AVPR2 gene; Gene mutation; Magnetic resonance imaging; Case report
lNTRODUCTlON
Congenital nephrogenic diabetes insipidus (CNDI) is a rare hereditary disease that accounts for approximately 10% of nephrogenic diabetes insipidus cases[1]. In approximately 90% of the cases, CNDI is caused by mutations in theAVPR2gene,which are inherited in an X-linked recessive manner. In the remaining 10%, CNDI is caused by mutations in the aquaporin-2 (AQP2) gene, which are inherited in an autosomal dominant or recessive manner[2,3].
Early clinical manifestations of CNDI in neonates are atypical. A lack of understanding of this disease by clinicians causes frequent misdiagnoses or missed diagnoses, which may result in failure to administer treatments in time, ultimately leading to serious complications. In this study, clinical data of a case ofAVPR2gene mutation-induced CNDI, which was confirmed by genetic testing, were retrospectively analyzed to improve our understanding of this disease.
CASE PRESENTATlON
Chief complaints
On February 1, 2020, a male neonate was hospitalized 17 d after birth due to a 7 d period of pyrexia.
History of present illness
Ten days after birth, the neonate developed pyrexia. The highest measured body temperature was 38.5 °C, and the neonate did not exhibit coughing, nasal congestion,rhinorrhea or rash and did not produce stools or urine with abnormal appearance. The neonate was presented at the First People’s Hospital of Shangqiu and was treated by infusion with cefoperazone-sulbactam. The patient remained to suffer from recurrent pyrexia and was therefore hospitalized at the Children’s Hospital Affiliated to Zhengzhou University for further diagnosis and treatment.
History of past illness
The patient was born with a birth weight of 3950 g at a gestational age of 40 + 1 wk.No abnormalities had been observed during prenatal examinations. No intrauterine distress or postnatal asphyxia had occurred.
Personal and family history
The parents were not consanguineous and were in good health. A family history of hereditary diseases or polydipsia and polyuria was negated.
Physical examination
Body temperature: 38.7 °C; pulse rate: 178 beats per minute; respiration rate: 56 breaths per minute; blood pressure: 90/67 mmHg (1 mmHg = 0.133 kPa); body weight: 4.08 kg; height: 55 cm; normal development, moderate nutritional status, clear consciousness, poor response, flat and soft anterior fontanel, skin and oral mucosa were mildly dry, normal skin elasticity, no resistance to neck flexion, no circumoral cyanosis; coarse respiration sounds in both lungs, a few coarse moist crackles, strong heart sounds, regular cardiac rhythm, soft abdomen, normal bowel sounds, normal muscle tone in all extremities and negative Kernig’s, Brudzinski’s and Babinski’s signs.
Laboratory examinations
Routine blood test + C reactive protein: white blood cell count: 12.92 × 109/L (high);red blood cell count: 4.11 × 1012/L; hemoglobin level: 133 g/L; platelet count: 300 × 109/L; neutrophil percentage: 60.4%; lymphocyte percentage: 25.4%; quantitative determination of C-reactive protein < 0.5 mg/L.
Routine urine tests: Urine specific gravity and osmolality were 1.005 (normal range:1.015-1.025) and 100 mOsm/kg (normal range: 550-1100 mOsm/kg), respectively;urine glucose: Negative; liver enzymes, renal function and myocardial enzymes:normal.
Electrolyte tests: Blood sodium level: 148.2-156 mmoL/L (normal range: l35-145 mmoL/L); blood chloride level: 110.7-122.8 mmoL/L (normal range: 95-107 mmoL/L); blood osmolality: 320 mOsm/kg (normal range: 275-305 mOsm/kg).
No abnormalities were observed in the cerebrospinal fluid. One month after discharge, results of genetic testing showed no abnormalities in theAQP2gene but revealed the hemizygous missense mutation c.541C>T (P.R181C) in theAVPR2gene(Figure 1). Genetic testing of the same locus in the patient’s family confirmed absence of mutations in the father (Figure 2) and presence of a heterozygous mutation in the mother (Figure 3).
Imaging examinations
No abnormalities were observed in the color doppler ultrasonographic imaging of the urinary system. Chest X-ray images revealed coarse textures in both lungs. Magnetic resonance imaging scans of the head and pituitary gland showed no abnormalities.
FlNAL DlAGNOSlS
Congenital nephrogenic diabetes insipidus due to the mutation inAVPR2(c.541C>T).
TREATMENT
Upon hospitalization, the patient was administered symptomatic supportive treatments, including meropenem for infection treatment, physical cooling and hypotonic solution to treat the electrolyte disturbance. Body temperature control was unsatisfactory with persistent pyrexia recurrence, and hypernatremia and hyperchloremia were difficult to correct. Monitoring of urine volume upon hospitalization showed urine volumes of 6-7.5 mL/(kg/h), which suggested diabetes insipidus, considering low urine specific gravity and high blood osmolality. Due to the patient’s age, water deprivation and vasopressin tests could not be completed successfully. Magnetic resonance imaging scans of the head and pituitary gland showed no abnormalities, which again supported a CNDI diagnosis. We observed mild dehydration in combination with mild dryness of the child’s skin and oral mucosa. The patient was fed on demand, and water was additionally provided between milk intakes (30-50 mL). A combination treatment of hydrochlorothiazide and amiloride was administered. After the treatment, dehydration was corrected, body temperature and electrolyte levels returned to normal, the volume of urine was significantly reduced and the patient was subsequently discharged.
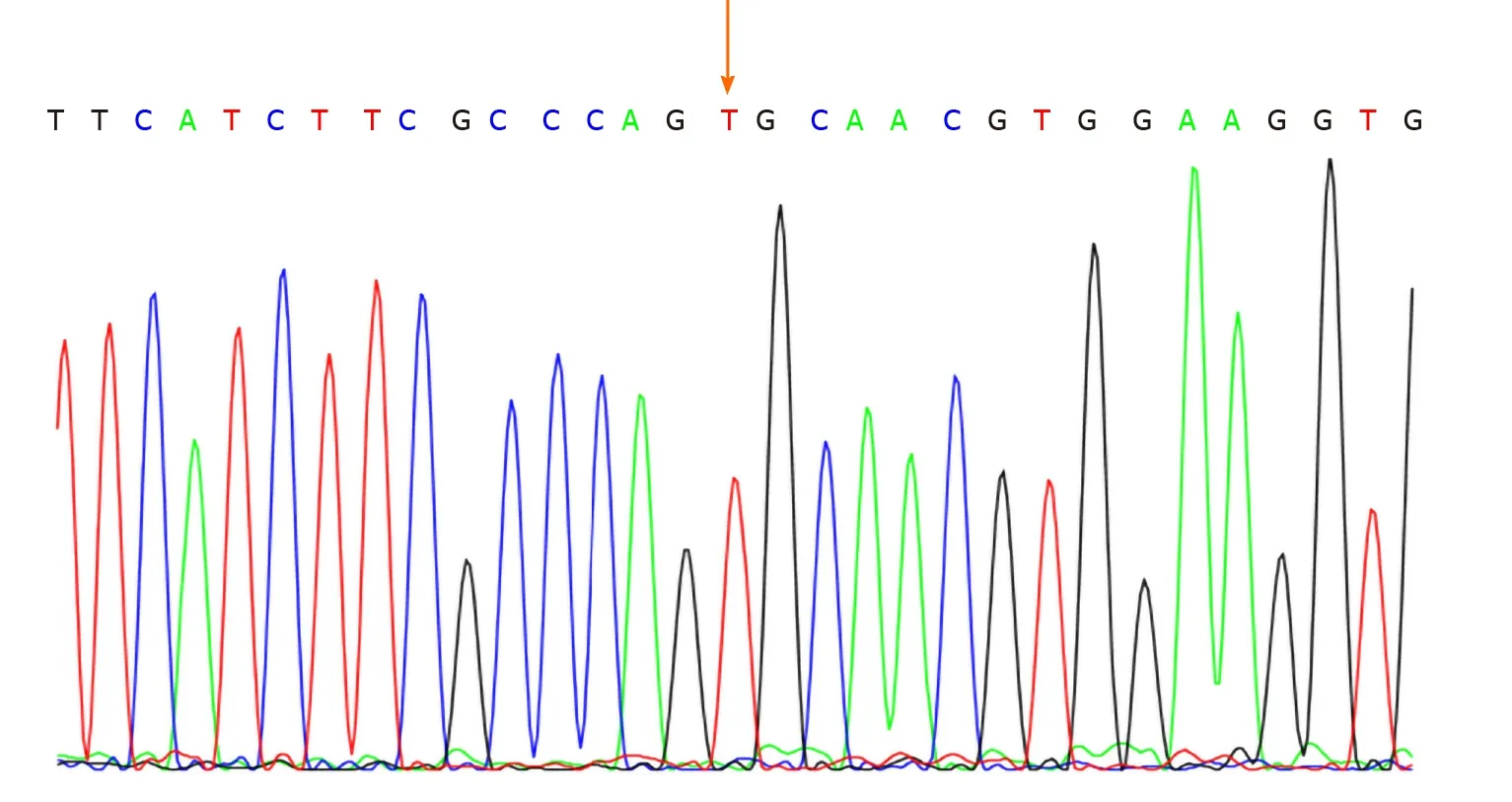
Figure 1 Genetic tests confirmed that the patient carried the AVPR2 gene mutation c.541C>T.
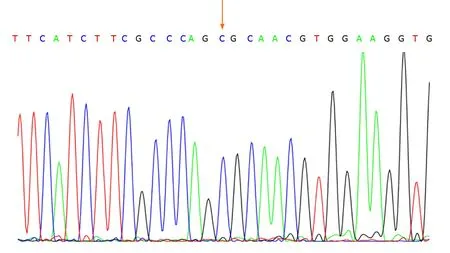
Figure 2 Genetic tests confirmed that the patient’s father did not carry the AVPR2 gene mutation c.541C>T.
OUTCOME AND FOLLOW-UP
After hospital discharge, the patient’s parents were instructed to breastfeed the neonate on demand and to supply sufficient water between milk intakes. Oral hydrochlorothiazide combined with amiloride was prescribed. Up until the most recent follow-up examination, normal body temperature, electrolyte levels and growth and development were observed.
DlSCUSSlON
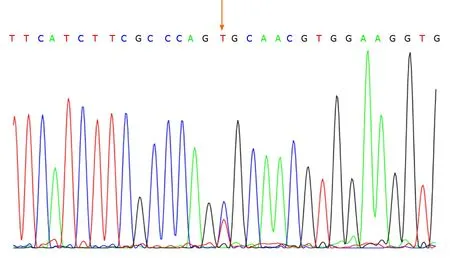
Figure 3 Genetic tests confirmed that the patient’s mother carried the AVPR2 gene mutation c.541C>T.
In this study, the presence of an X-linked inheritedAVPR2gene mutation on the Xchromosome was identified in a family. Mutations in the humanAVPR2gene were first reported by van den Ouwelandet al[4]in 1992. Under physiological conditions,arginine vasopressin (AVP) binds to AVPR2 receptors located on chief cells of the basement membrane, which coat the renal collecting ducts. This leads to activation of adenylate cyclase and produces cascading effects where protein kinase A is activated that in turn causes phosphorylation and subsequent translocation ofAQP2to the apical plasma membrane, resulting in water reabsorption and urine concentration[5].CNDI occurs when a site mutation in theAVPR2orAQP2gene causes insensitivity or unresponsiveness of the distal nephron to AVP, leading to urine concentration dysfunction and resulting in CNDI[6-8].
AVPR2gene mutation-induced CNDI accounts for approximately 90% of CNDI cases. The reported number of loci on theAVPR2gene where X-linked mutations may occur has gradually increased, and so far, more than 280AVPR2gene mutations have been identified. The majority of which are missense mutations[5,9]. In the present study,we identified the X-linked inherited missense mutation c.541C>T (P.R181C), which has not been reported previously in China.
Male patients with X-linked CNDI caused by mutations in theAVPR2gene are commonly encountered in clinical settings. By contrast, female patients with a heterozygous mutation exhibit different clinical phenotypes due to sex-biased X chromosome inactivation, which leads to various clinical manifestations and substantially different degrees of polydipsia and polyuria[9]. Early clinical manifestations of CNDI in neonates are atypical and include recurrent irritability,vomiting, feeding difficulties, slow body weight gain, hyperpyrexia, constipation and dehydration[10]. A lack of understanding of the disease by clinicians causes frequent misdiagnoses or missed diagnoses, which may result in failure to administer treatments in time and ultimately leads to severe complications such as mental retardation, growth retardation, hydronephrosis, renal insufficiency or serious electrolyte disturbance, which can be fatal[11,12]. Therefore, in cases of neonates exhibiting pyrexia of unknown cause accompanied by hypernatremia that is difficult to correct, diabetes insipidus should be considered when no significant abnormalities are found regarding infection indicators and when antibiotic treatments are ineffective.Urine volume and urine specific gravity should be monitored closely, and genetic screening should be conducted as soon as possible to facilitate a definitive diagnosis.Testing of theAVPR2andAQP2genes not only allows definitive diagnoses at an early stage but also facilitates timely treatment decisions, prevents serious complications such as mental retardation and developmental delay and provides information for familial cases and counseling regarding reproduction genetics[8].
CNDI is a hereditary disease caused by gene mutations. Thus no specific prevention measures are available[13]. Currently, symptomatic treatments targeting clinical manifestations related to urine concentration dysfunction are mainly adopted, such as adequate water supplementation, low-sodium diets and oral medications, so as to achieve symptom relief, prevent dehydration and hypernatremia, improve the quality of life and prolong life expectation[14]. Commonly used drugs include hydrochlorothiazide, amiloride and indomethacin. In the present case, oral treatment with hydrochlorothiazide and amiloride was started as soon as the diagnosis was established. Urine volume and laboratory parameters normalized, and a normal constant weight gain and a favorable psychomotor development have been observed.
Thiazides have long been used in these patients. Thiazide works by inhibiting reabsorption of sodium in the distal tubule by blocking the NaCl cotransporter, which leads to hypovolemia resulting in sodium and water reabsorption in the proximal tubules. Hydrochlorothiazide may also induce AQP2 upregulationviaan AVPindependent mechanism[15]. Thiazide diuretics in combination with the potassiumsparing diuretic amiloride helps reduce the degree of polyuria in patients with CNDI.Amiloride, a diuretic that blocks the epithelial Na channel along the cortical collecting tubule, provides additional natriuresis and can counteract thiazide-induced hypokalemia through its K-sparing effect. These drugs can relieve symptoms of diabetes insipidus to a certain extent but cannot cure the disease[16,17].
With the development of molecular genetic diagnostics and elucidation of the pathogenesis of CNDI, many novel treatment methods targeted towards the etiology of the disease have been proposed, suggesting that gene therapy may offer possibilities for curing CNDI. Therapeutic targets and strategies of the various methods differ between types of CNDI. At present, research on etiological treatment of CNDI is mainly focused on three aspects: (1) Reparation of misfolded proteins to restore normal protein structures that facilitates the release of proteins trapped in the endoplasmic reticulum and correct localization of proteins on cell membrane surfaces[17]; (2) Direct treatment of misfolded receptor proteins trapped in the endoplasmic reticulum[17]; and (3) Direct activation ofAQP2without mediation byAVPR2[1]. However, safety and efficacy of these methods for clinical application require further evaluation.
CONCLUSlON
CNDI in the neonatal period is rare, and its clinical manifestations are unspecific, with some patients merely showing recurrent fever and electrolyte disturbance. Genetic testing ofAVPR2andAQP2can be used for screening and genetic diagnosis of CNDI.Testing of theAVPR2andAQP2genes not only allows definitive diagnoses at an early stage but also facilitates timely treatment decisions, prevents serious complications such as mental retardation and developmental delay and provides information for familial cases and counseling regarding reproduction genetics.
 World Journal of Clinical Cases2020年24期
World Journal of Clinical Cases2020年24期
- World Journal of Clinical Cases的其它文章
- Primary duodenal tuberculosis misdiagnosed as tumor by imaging examination: A case report
- Successful endovascular treatment with long-term antibiotic therapy for infectious pseudoaneurysm due to Klebsiella pneumoniae: A case report
- Idiopathic adulthood ductopenia with elevated transaminase only: A case report
- Takotsubo cardiomyopathy associated with bronchoscopic operation: A case report
- Extracorporeal shock wave therapy treatment of painful hematoma in the calf: A case report
- Rare case of drain-site hernia after laparoscopic surgery and a novel strategy of prevention: A case report